Hallmarks of mechanochemistry: from nanoparticles to technology†
Peter Baláž*a, Marcela Achimovičováa, Matej Baláža, Peter Billikbc, Zara Cherkezova-Zhelevad, José Manuel Criadoe, Francesco Deloguf, Erika Dutkováa, Eric Gaffetg, Francisco José Gotore, Rakesh Kumarh, Ivan Mitovd, Tadej Rojaci, Mamoru Sennaj, Andrey Streletskiikl and Krystyna Wieczorek-Ciurowam
aInstitute of Geotechnics, Slovak Academy of Sciences, Watsonova 45, 04353, Košice, Slovakia. E-mail: balaz@saske.sk; Fax: +421557922604; Tel: +421557922603
bFaculty of Natural Sciences, Comenius University, Mlynská Dolina, 84215 Bratislava, Slovakia
cInstitute of Measurement Science, Slovak Academy of Sciences, Dúbravská cesta 9, 84104 Bratislava, Slovakia
dInstitute of Catalysis, Bulgarian Academy of Sciences, Acad. G. Bonchev St., Bldg. 11, 1113 Sofia, Bulgaria
eInstituto de Ciencia de Materiales de Sevilla, C.S.I.C., Américo Vespucio 49, 41092 Sevilla, Spain
fDipartimento di Ingegneria Meccanica, Chimica e dei Materiali, Università degli Studi di Cagliari, via Marengo 2, I-09123 Cagliari, Italy
gInstitut Jean Lamour, UMR 7198 CNRS, Ecole des Mines de Nancy, Université de Lorraine, Parc de Saurupt – CS 14234, F54042 – Nancy Cedex, France
hCSIR-National Metallurgical Laboratory, Jamshedpur-831 007, India
iJožef Stefan Institute, Jamova cesta 39, 1000 Ljubljana, Slovenia
jFaculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, 223-8522, Japan
kInstitute Chemical Physics, Russian Academy of Sciences, Kosygina 4, 119991 Moscow, Russia
lMoscow Institute of Physics and Technology, 9 Institute str., Dolgoprudniy, Moscow region, 141700, Russia
mFaculty of Chemical Engineering and Technology, Cracow University of Technology, 24 Warszawska Str., 31-155 Cracow, Poland
First published on the web 4th April 2013
The aim of this review article on recent developments of mechanochemistry (nowadays established as a part of chemistry) is to provide a comprehensive overview of advances achieved in the field of atomistic processes, phase transformations, simple and multicomponent nanosystems and peculiarities of mechanochemical reactions. Industrial aspects with successful penetration into fields like materials engineering, heterogeneous catalysis and extractive metallurgy are also reviewed. The hallmarks of mechanochemistry include influencing reactivity of solids by the presence of solid-state defects, interphases and relaxation phenomena, enabling processes to take place under non-equilibrium conditions, creating a well-crystallized core of nanoparticles with disordered near-surface shell regions and performing simple dry time-convenient one-step syntheses. Underlying these hallmarks are technological consequences like preparing new nanomaterials with the desired properties or producing these materials in a reproducible way with high yield and under simple and easy operating conditions. The last but not least hallmark is enabling work under environmentally friendly and essentially waste-free conditions (822 references).
 From left to right: (top row) Peter Baláž, Marcela Achimovičová, Matej Baláž, Peter Billik, Zara Cherkezova-Zheleva, José Manuel Criado, Francesco Delogu, Erika Dutková; (bottom row) Eric Gaffet, Francisco José Gotor, Rakesh Kumar, Ivan Mitov, Tadej Rojac, Mamoru Senna, Andrey Streletskii, Krystyna Wieczorek-Ciurowa | Professor Peter Baláž graduated in chemistry at the Faculty of Natural Sciences, P.J. Šafárik University, Košice, in 1971. He has been active in the field of mechanochemistry since 1977 and his basic and applied research encompasses the field of solid-state chemistry, nanoscience and minerals engineering. He is the author of 4 books and 240 papers in reviewed journals. His papers have been cited more than 1100 times in Science Citation Index. He is a founding member of the International Mechanochemical Associaton. Since 1977 he has worked at the Institute of Geotechnics of Slovak Academy of Sciences in Košice, Slovakia. |
1. Introduction
1.1 Definitions
Despite the long history of mechanochemistry, the definition of a mechanochemical reaction was only recently incorporated into the chemical literature. The recent IUPAC Compendium of Chemical Terminology defines mechanochemical reaction as a “chemical reaction that is induced by mechanical energy”.1 However, the term mechanochemistry was introduced by Ostwald2a,b much earlier. He was engaged in the systematization of chemical sciences from the energetic point of view. He understood mechanochemistry in a wider sense when compared with the present view, regarding it as a part of physical chemistry at the same level as thermochemistry, electrochemistry or photochemistry. After ten years, the practically forgotten book by Pierce on mechanochemistry was published.2c He defined mechanochemistry as “the new science of mechanical dispersion involving the use of principles in physical chemistry”. He hesitated to name this science, remarking “we shall call it mechanochemistry for lack of a better name, as it involves dispersion or defflocculation by mechanical means, thereby bringing about so-called colloidal dispersions”. In this book, the relationship between mechanochemistry and nanoscience can be traced for the first time (colloidal dimensions = 1–1000 nanometers) and the first colloidal mill (Fig. 1) was advertised there. Subsequently, the boundaries of mechanochemistry contracted. For instance, Hüttig2d assumes that mechanochemistry includes only the release of lattice bonds without any formation of new substances (i.e. he supports the physical approach), while Peters2e also puts transformations due to mechanical stress of material that are accompanied by chemical reaction into this category. It was Butyagin2f who contributed a certain unification. He considered the behaviour of solids exposed to the effect of mechanical energy from the viewpoint of three main aspects: structural disordering, structure relaxation and structural mobility. In real conditions, these three factors simultaneously affect the reactivity of solids.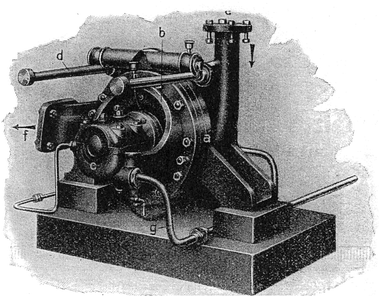 | ||
| Fig. 1 Plauson-Oderberg colloid mill for wet milling.3 | ||
At present, the definition of Heinicke2g is widely accepted: “mechanochemistry is a branch of chemistry which is concerned with chemical and physico-chemical transformations of substances in all states of aggregation produced by the effect of mechanical energy”.
There is another frequently used term in mechanochemistry – mechanical activation (MA). The term was introduced by Smékal,4a who regarded it as a process involving an increase in reaction ability of a substance which remains chemically unchanged. In this case, the MA precedes the reaction and has no effect during the course of this reaction. Provided the activation brings about a change in composition or structure, it is a mechanochemical reaction. The definitions of mechanical activation published later were always dependent on the observed effect. Butyagin4b defined MA as an increase in reaction ability due to stable changes in solid structure. Structural relaxation plays an important role in mechanical activation. The concept of slowly changing states after interrupting the action of mechanical forces has been described by Lyachov.4c He published a generalised relaxation curve for activated solids where individual parts of the curve correspond to processes with different characteristic times of relaxation (Fig. 2).
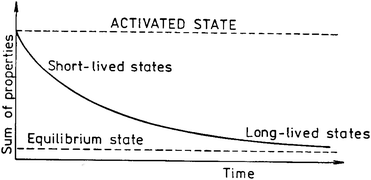 | ||
| Fig. 2 A generalised relaxation curve of a mechanically activated state.4c | ||
By this theory there is no possibility of influencing the reactivity of activated solids in states whose relaxation times are shorter than the characteristic time of the reaction itself. On the contrary, some long-living states (e.g. surface area) may be regarded as constant during the course of reaction and their influence has to be a subject of mechanical activation studies. As for the kinds of relaxation processes, various processes were described: heating, formation of a new surface, aggregation, recombination, adsorption, imperfections, chemical reaction between adjoining particles, etc.4d,e The rate of these relaxation processes may be vastly different and the processes can change from one way of relaxation to the other. Thus, MA can be regarded as a multi-step process with changes in the energetic parameters and the amount of accumulated energy of solids in each step. The four processes, namely the accumulation of defects, amorphization, the formation of metastable polymorphous forms and chemical reaction, are united by the term mechanical activation. Especially, various types of defects (Fig. 3) play important roles in mechanochemistry.4f
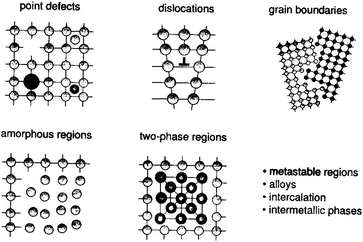 | ||
| Fig. 3 Defects created by MA of solids. Reprinted with permission from ref. 4g. Copyright 2005, Wiley. | ||
Juhász proposed that processes under the influence of mechanical activation can be subdivided into primary and secondary ones.4e,h,i The primary process (e.g. increase of internal and surface energy, increase of surface area, decrease of the coherence energy of solids) generally increase the reactivity of the substance. The secondary processes (e.g. aggregation, adsorption, recrystallization) take place spontaneously in activated systems and may appear even during milling or after milling has been completed.
However, the terms mechanochemistry and mechanical activation (MA) are not the only ones. Even contributors to this critical review use different terminology. For this reason a list of abbrevations has been inserted at the end of this paper.
1.2 History of mechanochemistry
Important players. There are several approaches on how to deal with this topic. According to the paper by Takacs,5a the earliest documented mechanochemical reaction may have been milling of cinnabar (HgS). Theophrastus of Eresus (371–286 BC), student and successor of Aristotle at the Lyceum in Athens, wrote in his book De Lapidibus (On Stones) that “native cinnabar was rubbed with vinegar in a copper mortar with a copper pestle yielding the liquid metal”. This is a very clear description of a mechanochemical process. The first described mechanochemical reduction probably followed the reaction
| HgS + Cu → Hg + CuS | (1) |
where vinegar was added to prevent the side effects which usually accompany dry milling in air. It remains a mystery why the mechanochemical preparation of mercury from its sulphide according to reaction (1) was forgotten during the Middle Ages. However, as published in ref. 5a, examples of other mechanochemical reactions between 300 BC and the end of the 18th century can be also traced in medieval literature. Agricola documented several examples of chemical reactions under the influence of mechanical action which can be connected with mining and metallurgical operations.5b,c It is interesting to note that in the 17th century Bacon referred to four treatments that, in essence, are still among the most important procedures to prepare active solids. One of them is milling.5d It was Wenzel who stressed the fact that the degree of conversion of heterogeneous reactions depends mainly on the surface area of the reacting solids and is not proportional to their amount.5e
The quick look into the history of mechanochemistry in ancient and medieval times above is not exhaustive. Many other important scientists contributed to the development of mechanochemistry, e.g. Baramboin, Bowden, Carey Lea, Clark, Dachille, Faraday, Flavickii, Fink, Fox, Grohn, Gutmann, Heinicke, Hofman, Hüttig, Juhász, Khodakov, Oprea, Ostwald, Parker, Paudert, Peters, Rowan, Roy, Schrader, Simoniescu, Smékal, Tabor, Tamman, Thiessen, Yoffe and Wanetig. This list of mechanochemists is not complete but all of them contributed to the establishment of mechanochemistry.6,7 The small (and incomplete) excursion into history of mechanochemistry can be broadened by reading several review papers given in Table 1.
Table 1 Review papers on history of mechanochemistry
| Title | Author(s) and year | Reference |
|---|---|---|
| Mechanochemische reaktionen | Peters, 1962 | 2d |
| Review of the phase transformation and synthesis of inorganic solids obtained by mechanical treatment (mechanochemical reactions) | Lin and Nadiv, 1970 | 8a |
| Mechanically initiated chemical reactions in solids | Fox, 1975 | 8b |
| Mechanochemistry of inorganic solids | Boldyrev, 1986 | 4d |
| Accelerating the kinetics of low-temperature inorganic synthesis | Roy, 1994 | 8c |
| Colloid-chemical aspects of mechanical activation | Juhász, 1998 | 4j |
| Mechanochemistry of solids: past, present and prospects | Boldyrev and Tkáčová, 2000 | 4f |
| Mechanochemistry in extractive metallurgy: the modern science with old routes | Baláž, 2001 | 8d |
| M. Carey Lea, the first mechanochemist | Takacs, 2004 | 8e |
| Mechanochemistry: the mechanical activation of covalent bonds | Beyer and Clausen-Schaumann, 2005 | 8f |
| Mechanochemistry and mechanical activation of solids | Boldyrev, 2006 | 8g |
| The mechanochemical reduction of AgCl with metals: revisiting an experiment of M. Faraday | Takacs, 2007 | 8h |
In the modern history of mechanochemistry the International Conference on Mechanochemistry and Mechanical Alloying (INCOME) series was initiated by the International Mechanochemistry Association IMA (an associate member of IUPAC). IMA was established in 1988 in Tatranská Lomnica, Slovakia, see Fig. 4. The first INCOME conference was held in Košice (1993) and it was followed by conferences in Novosibirsk (1997), Prague (2000), Braunschweig (2003), Novosibirsk (2006), Jamshedpur (2008) and Herceg Novi (2011).
 | ||
| Fig. 4 The foundation of International Mechanochemical Association (IMA) in Tatranská Lomnica, Slovakia, in 1988. From left to the right: A. P. Purga (Russia), V. Jesenák (Slovakia), I. Hocmanová (Slovakia), L. G. Austin (USA), P. Baláž (Slovakia), M. Senna (Japan), interpreter, E. G. Avvakumov (Russia), L. Opoczky (Hungary), K. Tkáčová (Slovakia), N. Z. Lyachov (Russia), V. V. Boldyrev (Russia), H.-P. Hennig (Germany), N. Števulová (Slovakia), H.-P. Heegn (Germany), P. Yu. Butyagin (Russia). | ||
Theories and models. Several theories and models in the history of mechanochemistry have been elaborated.6 In hot-spot theory, the hypothesis for the reason of mechanical initiation of chemical reactions was developed by Bowden, Tabor and Yoffe.9a–c They found out that with friction processes, temperatures of over 1000 K on surfaces of about 1 μm2 can occur and last for 10−4–10−3 s and that they represent an important cause of mechanically initiated reactions. These temperatures can be also reached near the tip of a propagating crack.9d Later this theory was expanded for other processes, e.g. oxidation of metals. Several interpretations have been described.7 For example, it was proved that the processes occurring at the tip of a crack during the cleavage of a crystal may proceed by different mechanisms depending on the speed of crack motion.9e,f This theory has been strongly criticized recently.9g In the sixties, the first model in mechanochemistry – the magma-plasma model – was proposed.10 According to this model, a huge amount of energy is set free at the contact spot of colliding particles. This energy is responsible for the formation of a special plasmatic state which is characterized by emission of fairly excited fragments of solid substance, electrons and photons over a short time (Fig. 5). The surface of colliding particles is rather disordered and local temperatures can reach more than 10![[thin space (1/6-em)]](./Hallmarks_files/char_2009.gif) 000 K. Thiessen et al. distinguish the reactions which occur in the plasma from the reactions taking place at the surface of particles during the significantly excited state or immediately after its expiration. These considerations led to an important conclusion which is valid for mechanically activated reactions: these reactions do not obey a single mechanism.
000 K. Thiessen et al. distinguish the reactions which occur in the plasma from the reactions taking place at the surface of particles during the significantly excited state or immediately after its expiration. These considerations led to an important conclusion which is valid for mechanically activated reactions: these reactions do not obey a single mechanism.
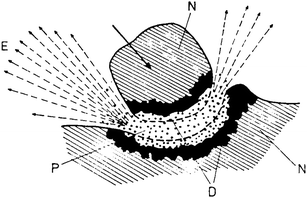 | ||
| Fig. 5 Magma-plasma model: E – exo-electrons, N – undeformed solid, D – highly deformed surface layer, P – plasma.10 | ||
Later on, mainly German and Russian mechanochemists have developed several other models and theories like the spherical model,2fdislocation and phonon theory,11a,b the theory of short-living active centers,11c the kinetic model11d (expanded recently11e,f), the impulse model4d,11d,g and the analogy model.11h These models and theories are described in more details in a monograph (ref. 7).
1.3 Mechanochemical tools and how to rule them
The multi-stage character of MA requires high-energy mills with different working regimes (compression, shear, impact). The principles of the most frequently applied mills are shown in Fig. 6.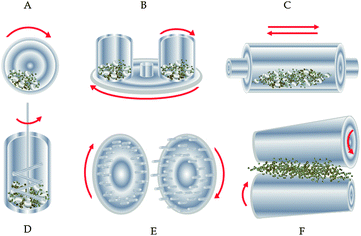 | ||
| Fig. 6 Types of mills for high-energy milling: A – ball mill, B – planetary mill, C – vibration mill, D – attritor (stirring ball mill), E – pin mill, F – rolling mill.4k Modified from ref. 4d. | ||
There are several variables which influence the milling process, e.g. type of the mill, material of milling media, ball-to-powder ratio, filling extent of the milling chamber, milling atmosphere, milling speed, milling time, etc.7
The purpose of an ideal device for a mechanochemical synthesis (MCS) is to insert the maximum amount of energy into the treated solid to enable the accumulation of the input energy. This energy is responsible for the occurrence of defects, which greatly affect chemical reactivity. This requires high-energy inputs to be transferred from the working medium to the treated solid during the mechanochemical reaction. Due to their high energy density, and simple set-up, handling and cleanability, planetary ball mills (for example, manufactured by Fritsch or Retsch, Germany) are especially suitable for various MCS processes.12–17 At the laboratory scale, the MCS can be realized in a disc vibration mill as well.18 Industrial planetary mills with continuous action, characterized by productivity of up to 3–5 tons per hour, are now commercially available. The energy density in these mills is 100–1000 times higher than the energy density used in earlier conventional milling equipment.19 Mechanochemical vials or reactors and balls are available in different materials such as agate (SiO2), silicon nitride, sintered corundum, zirconia (tetragonal ZrO2 stabilized with Y2O3), chrome steel, Cr-Ni steel, tungsten carbide, or polyamide.14,15 The ball-to-powder ratio (BPR) (weight of the balls to the powder) has been varied by different investigators from a value as low as 1:1 to as high as ![[similar]](./Hallmarks_files/char_223c.gif) 200:1. The ratio of 10:1 is most commonly used in small capacity mills such as a SPEX mill.14 Supplementary technical details of mechanochemical parameters such as the type of material of the milling bodies and vials, ball filling ratio, milling atmosphere, processing control, organic agents, milling speed, etc. can be found in ref. 12, 14 and 15, and useful contributions to details and techniques to determine and calculate the physics, kinetics, and energetics between milling media during high-energy milling (HEM) can be found in ref. 12 and 20–22. The need to use a protective inert atmosphere in the case of moisture- or oxygen-sensitive materials has been discussed in many reviews.14,23–25 In such a case, the vial is usually charged and purged in a glove box, which is rather cumbersome. An alternative would be the development of devices that allow operating the mill while it is permanently connected to the gas container. Ogino et al.26 and Ching and Perng27 measured the pressure in a gas container that was connected to the milling vial by means of a plastic tube. They used this method to study the kinetics of the mechanochemical reaction between titanium and nitrogen. The kinetics of mechanochemical hydrogenation of metal alloys with potential applications as hydrogen storage materials has been also studied by several authors28–32 with similar procedures. Shaker mills were used in all of the mentioned works. A procedure that allows the operation of a planetary mill with the vial connected to a gas cylinder has been also described.33 The vial and the gas cylinder were connected by a flexible polyamide tube through a rotary valve to avoid the problem of the spinning movement of the connecting tube. This system has been successfully used for the mechanochemical synthesis of refractory nitrides and carbonitrides under nitrogen pressures up to 10 bar.34,35
200:1. The ratio of 10:1 is most commonly used in small capacity mills such as a SPEX mill.14 Supplementary technical details of mechanochemical parameters such as the type of material of the milling bodies and vials, ball filling ratio, milling atmosphere, processing control, organic agents, milling speed, etc. can be found in ref. 12, 14 and 15, and useful contributions to details and techniques to determine and calculate the physics, kinetics, and energetics between milling media during high-energy milling (HEM) can be found in ref. 12 and 20–22. The need to use a protective inert atmosphere in the case of moisture- or oxygen-sensitive materials has been discussed in many reviews.14,23–25 In such a case, the vial is usually charged and purged in a glove box, which is rather cumbersome. An alternative would be the development of devices that allow operating the mill while it is permanently connected to the gas container. Ogino et al.26 and Ching and Perng27 measured the pressure in a gas container that was connected to the milling vial by means of a plastic tube. They used this method to study the kinetics of the mechanochemical reaction between titanium and nitrogen. The kinetics of mechanochemical hydrogenation of metal alloys with potential applications as hydrogen storage materials has been also studied by several authors28–32 with similar procedures. Shaker mills were used in all of the mentioned works. A procedure that allows the operation of a planetary mill with the vial connected to a gas cylinder has been also described.33 The vial and the gas cylinder were connected by a flexible polyamide tube through a rotary valve to avoid the problem of the spinning movement of the connecting tube. This system has been successfully used for the mechanochemical synthesis of refractory nitrides and carbonitrides under nitrogen pressures up to 10 bar.34,35
1.4 Accompanying phenomena
Several typical phenomena connected with MA and mechanochemical synthesis (MCS) processes can be mentioned here.7,14 Since diffusion processes are involved in the formation of a nanostructure, it is expected that the temperature of milling will have a significant effect. Two kinds of temperature effects during milling are usually taken into account: local temperature pulses due to ball collisions and the overall temperature in the vial.14 The temperature increase of the milling balls in two laboratory mills was studied recently.36 The ball temperature remains below 100 °C in a SPEX mixer mill. Temperatures over 200 °C are typical for a planetary mill operating at similar milling intensities. A temperature decrease is expected at very high speeds, as the balls stay attached to the vial wall for longer, reducing both heating and efficiency of milling.37A serious problem that usually occurs in mechanochemical research is contamination. The small size of milled particles, the availability of large surface area and the formation of new surfaces during milling all contribute to the contamination of the powder.14 Using a “seasoned” milling vial (i.e. media coated with the product powder) resulted in very low values for Fe contamination.38
During high-energy milling, the size of crystals decreases to some critical value. Further energy supply to these crystals of limiting size causes further deformation of crystals, energy accumulation in the volume or at the surface of crystals, and subsequently amorphization.4f
The particle size reduction is in many cases complicated by particle size enlargement, where smaller particles are put together to form larger entities in which the original particles can still be identified. This phenomenon, called equilibrium state of milling, was experienced with solids and is closely related to the effects of aggregation and agglomeration.
In the course of the milling process, a gradual deterioration of effectiveness is observed. Thorough investigation of this process on several solids has shown that three stages can be clearly distinguished:39a–c the Rittinger stage (a), in which the interaction of particles can be neglected; the aggregation stage (b), in which the new surface area produced is not proportional to the energy input because of particle interaction (aggregation); and the agglomeration stage (c), in which the increase of dispersion first drops to a negligible value and then stops altogether. While in stage (b) the aggregates are mechanically connected via van der Waals forces of magnitude 0.04–4 kJ mol−1, in stage (c) the particles are kept together by chemical bonds of magnitude 40–400 kJ mol−1. Mechanochemical reactions and changes in the crystal structure mainly occur at this stage. In Fig. 7 the characteristic plot is given.
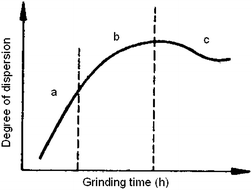 | ||
| Fig. 7 Three stages during high-energy milling of solids. Reprinted with permission from ref. 39a. Copyright 1977, Elsevier. | ||
2. Theoretical aspects
In contradiction with older definitions (see section 1), mechanochemistry can be also defined as the chemistry in which the change of the thermodynamic state variables and functions of a given chemical system, including at least one solid phase, can be ascribed to the effects of non-hydrostatic mechanical stresses, and of the resulting plastic strain.40,41 Although the Onsager’s reciprocal relations suggest that thermal and mechanical effects can be hardly separated,40,42 two important differences between thermally and mechanically activated processes can be readily pointed out. First, mechanochemical processes invariably take place under non-equilibrium conditions, the chemical reactivity being promoted by unbalanced mechanical forces.41,43 Second, whereas temperature is an intensive thermodynamic state variable, mechanical deformation occurs on a local basis, being mediated by dislocations and other lattice defects.44Based on these premises, it must be expected that the mechanical activation induces mass transport processes different from the ones operating under thermal activation conditions, with a significant impact on the physical and chemical behavior of solids at different scales.
2.1 Atomistic processes
Being connected with mechanical deformation, mechanochemical transformations exhibit a marked local character. Whereas this complicates experimental investigations, it makes their study amenable to numerical simulations. Although with the unavoidable limitations of system size and simulation time length, molecular dynamics methods have thrown some light on the atomistic processes occurring in response to localized mechanical stresses, particularly in the case of metallic phases.The first studies focused on the behavior of surfaces and interfaces in open dissipative systems in which an external forcing, typically a sustained shearing, acts in competition with thermally activated relaxation.45,46 Under such conditions, smooth interfaces exhibit a considerable kinetic instability. A fine-grained microstructure is developed due to an intense dislocation activity, which also promotes chemical mixing.47,48 This is mediated by the intermittent rotation of small clusters of atoms, shown in Fig. 8, which provides an atomic displacement mechanism alternative to thermal diffusion.47–51
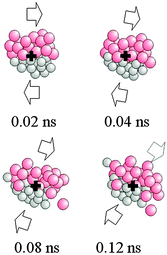 | ||
| Fig. 8 The rotation of a small atomic cluster. | ||
The dynamics of atomic displacements is affected by both system geometry and shearing intensity.52–55 In the case of smooth interfaces, the distance covered by displacing atoms exhibits a dependence on the square root of time similar to the one of thermal diffusion.47–51 Conversely, in composite precipitate-matrix systems, the dependence on time approaches the one expected for turbulent flows.52,53 Atomic mobility is also affected by the heat of mixing54 and the mechanical properties.53,55 In particular, a superdiffusive behavior is observed when the dislocations moving in response to the external forcing are able to cross the interfaces separating different chemical species.55
The severity of local mechanical deformation increases when sliding is replaced by a frictional collision regime.56–59 The deposition of kinetic energy at rough surfaces initiates a complex sequence of atomistic processes resulting in the local excitation of about 200 to 1000 atoms, which experience instantaneous temperatures as high as 1000 or 2000 K for time intervals of about 2 to 10 ns.56,57 The interaction of surface asperities with each other during the first collision stages (shown in Fig. 9) generates liquid-like regions characterized by instantaneous temperatures and pressures of about 10000 K and 17 Gpa, respectively.58 Under such conditions, unexpected alloying mechanisms based on material transfer operate. Nanometer-sized domains of the softer chemical species are transported to the surface of the harder one. Due to the reduced size, such domains can melt at temperatures lower than the equilibrium one, thus inducing the dissolution of the harder chemical species and the subsequent alloying.59
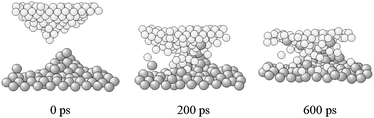 | ||
| Fig. 9 The frictional collision between two surface asperities. | ||
Interesting per se,60 material transfer processes are sensitive to the shape of surface asperities, to the work of adhesion, and to the mutual orientation of crystallographic planes.61–63 Thresholds in both the intensity of mechanical stresses and the work of adhesion can be identified, below which no mechanical deformation or no material transfer take place. Above the, the material behavior can be rationalized in terms of models related to the nucleation of dislocations at the crack tip. It follows that the overall response to sliding contact is also modulated by temperature and mechanical properties;61–63 these are also progressively modified due to the hardening induced by the interaction of surfaces.64,65
In highly exothermic systems, the severe mechanical deformation occurring at the points of contact between surface asperities can give rise to a relatively complex chemical behavior. The deposition of kinetic energy, accompanied by the generation of lattice defects and the rise of temperature, promotes local melting phenomena. Due to the large negative enthalpy of mixing, the subsequent mutual dissolution of chemical species induces a marked temperature increase, which finally initiates a self-propagating chemical reaction.66
Although the conceptual framework is still fragmentary, the above-mentioned numerical findings suggest for mechanically activated processes a complex, multistage mechanism involving forced atomic displacements under the effects of mechanical stresses, and a subsequent thermally activated relaxation. This mechanism only accounts for the fundamental processes underlying the mechanochemical transformation, which also involves phenomena on coarser scales.
2.2 Friction, wear, and fracture
On the macroscopic scale, the substantial interaction of surface asperities results in friction phenomena, i.e. a resistance to the relative motion of the solid surfaces.67 On the mesoscopic scale, friction is associated with wear, i.e. the displacement of material on a solid surface induced by the mechanical stresses operating when sliding.68 Occurring when surfaces slide against each other for relatively long distances, wear cannot be comprehensively studied by numerical simulations. Rather, these can be used as a supplement to direct experimental investigations.69–73The frictional sliding of solid surfaces gives rise to large plastic strains and strain gradients as well as to high strain rates and strain rate gradients. In turn, these induce deep structural changes in the regions adjacent to the sliding interface, also promoting far-from-equilibrium chemical reactivity.69–73
According to molecular dynamics simulations, sliding can drive mass flows similar to the ones observed in fluids.71–74 So-called Kelvin–Helmholtz shear instabilities emerge, giving rise to local vorticity,74 as shown in Fig. 10. Vortices exhibit the same size scale of individual grains in nanostructured phases and can be associated with both chemical mixing and the formation of the nanostructure under dynamic deformation conditions.69–74
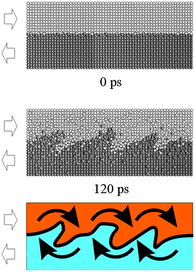 | ||
| Fig. 10 Vorticity due to the emergence of the Kelvin–Helmholtz shear instability at the sliding interface. | ||
The deposition of kinetic energy accompanying the mechanical deformation not only promotes the refinement of the system microstructure, but also abrasion and fracture.75–77 Due to its importance for human activities, fracture has been widely studied, particularly with the aim of identifying criteria for the nucleation and propagation of cracks.76,77 Conversely, its impact on the mechanochemistry of materials has been only scarcely investigated.
Determining an increase of the specific surface area, fracture has a definite effect on the rate of heterogeneous mechanochemical processes, enhancing the probability of contact between the different reactants.78–81 This extensive effect can be also associated with an inherently mechanochemical one.
A few attempts to characterize the mechanically activated chemistry at interfaces were carried out in the past, focusing on the formation of dangling bonds and related phenomena at the surface of covalent and metallic solids in sliding contact.82,83 Regarding the new surfaces generated by fracture, numerical findings suggest that they can exhibit a marked chemical reactivity due to the chemical bond unsaturation. However, the chemical reactivity is rapidly quenched by the relaxation processes, resulting in surface reconstruction and chemical reaction with the environment.84–88 As a consequence, the experimental study of the mechanochemical processes associated with fracture is quite difficult.
Despite this, chemical reactions have been successfully used to indirectly probe, on a very short time scale, the reactivity of the surfaces generated by fracture. In particular, it has been shown that the active sites located at the surface of fractured quartz particles interact with ethanol molecules to generate hydrogen radicals, which react in turn with a radical scavenger.89 The kinetics of the scavenger consumption suggests that the active sites exhibit enhanced chemical reactivity, which raises their apparent surface density above the maximum possible number of dangling bonds per surface unit.89 Accordingly, the surface formed by fracture exhibits a reactivity higher than the one that can be simply ascribed to a steady-state population of dangling bonds.
As a whole, the experimental and theoretical findings indicate that the surfaces generated by fracture, and the regions affected by severe deformation, undergo fast dramatic changes once the fracture has occurred or the mechanical stresses have been removed. These changes irreversibly quench the unusual system reactivity exhibited under dynamic conditions, which specifically relates to mechanochemistry. Understanding the processes underlying such reactivity represents one of the fundamental challenges for the future of mechanochemistry.
Local atomic processes, mechanical deformation, fracture, and specific surface area increase occur simultaneously to define the kinetics of mechanochemical transformations.
2.3 Kinetics
The kinetics of processes where mechanical activation takes place have been widely studied in the past for a variety of heterogeneous systems including inorganic solids.16,90–96 In most cases, the kinetic study was approached along the perspective of classical chemical kinetics, which relies upon thermally activated processes taking place continuously in time.97 Within this framework, the external forcing was described as a perturbation of the thermal dynamics, and the surface area increase superposed to the overall kinetic scheme. This led to the definition of kinetic laws qualitatively similar to the ones of classical chemical kinetics, but with characteristic parameters quantitatively modulated by the intensity of the external forcing.16,90–96 In ref. 91, an attempt to apply principles of solid-state heterogeneous kinetics on mechanochemical systems has been described. However, only a few common features with “classical” heterogeneous kinetics can be found (e.g. solid-state diffusion) and therefore special approaches accepting the peculiarities of mechanochemistry are needed. Due to the impulse character, space non-uniformity and changes of the conditions for chemical interactions during a mechanochemical process, its kinetic description is a complicated task. Therefore, there is no general approach possible and only some particular models are feasible.Since the dispersing process and mechanical activation develop as statistically probable processes, it is possible to consider chemical interaction from the viewpoint of collision theory. In this case, group collisions of surface atoms of two different particles in the zone of mechanical action are considered. The limiting stage of this process is the probability of formation of contacting regions between solid particles of the components and a collision with a milling body.16 According to the mentioned statements, the rate of reaction in a mill can be estimated as
| v = KmxSn | (2) |
where Km is a constant characterizing the probability of reaction to occur at a given mechanical action per unit contact (energy constant determining thermodynamic reaction parameters T and p), x is a probability of contacting particles to come across a collision with milling body, Sn is an area of contacting regions of A and B components during mechanical action.91 Dispersion brings about an increase of Sn, since the surface area of interacting components increases. The process of new surface area formation can be described by the equation
| S = Sm(1 − e−kt) | (3) |
where S is the specific surface area after certain time of milling t and Sm is the maximum specific surface area.98 Constant k implies the significance of the rate constant of new surface formation. Eqn (3) describes the processes in which the formation of a new surface is limited by the milling equilibrium after a certain time of milling.
Although successful in the definition of phenomenological rate laws able to interpolate experimental data, such an approach is no longer satisfactory when the experimental findings are used to gain indirect information on the atomic scale processes governing the mechanochemical transformation. First, it does not take into account the far-from-equilibrium character of mechanochemistry.99 Second, it does not relate, on a fundamental basis, the chemical reactivity to the severity of the mechanical deformation.100 Third, it does not connect the overall kinetics with the characteristic features of the mechanical processing method.101
Regarding the first issue, it must be noted that any novel achievement in such a direction would be significant progress in fundamental knowledge in the field. Unfortunately, only a few attempts have been made,99 and the results obtained are not sufficient to define the theoretical framework.
Conversely, significant progress has been made in the development of a conceptual framework able to describe mechanochemical transformations starting from a coherent set of assumptions. The theoretical approach focuses on the experimental case of so-called high-pressure torsion.102–105 High-pressure torsion is a severe mechanical deformation method that exploits the axial rotation of two Bridgman’s anvils. It allows study of the material behaviour under high pressure and large plastic strain conditions.106 The combination of large plastic shear with high pressure drastically modifies the structure and the properties of materials, as well as the thermodynamics and kinetics of their physical and chemical transformations. For example, the pressure at which phase transitions occur is reduced, and the kinetics can be described as a function of plastic strain, since time loses its usual relevance.102–106 The use of transparent diamond anvil cells represents a considerable breakthrough in the field. It allows collection of experimental data on the structure of the processed material by diffraction of synchrotron X-ray radiation. Therefore, experimental findings and theoretical predictions can be suitably compared.102–105 The nucleation of new lattice defects being related to the plastic flow, structural changes are related to plastic strain, rather than to mechanical stresses. The strain is able to reduce the pressure at which a given structural change takes place, and a barrier-less nucleation can occur at high concentrations of defects. Under such circumstances, the kinetics is intrinsically dominated by plastic strain, and time can no longer be the reference quantity. Furthermore, the competition between thermally activated recovery processes and strain-induced mechanochemical processes does not allow complete conversion, but rather a partial transformation characterized by the achievement of steady states depending on the total pressure.102–105
Far from being exhaustive and satisfactory, the studies regarding high-pressure torsion indicate that strain-induced transformations require a completely different thermodynamic and kinetic description. Understanding the principles and the mechanisms underlying the effect of plastic shear on mechanochemical transformations is extremely important from the point of view of fundamental physics, as well as for the identification of innovative materials and the improvement of the experimental methods for their synthesis.
In this regard, it must be noted that the above-mentioned theoretical approach to mechanochemistry is not restricted to the specific case of high-pressure torsion. On the contrary, a recent experimental study has pointed out the possible unexpected connection between high-pressure torsion and the mechanical processing of powders by ball milling.107 Based on a set of experimental findings, including total amount of final product, average size of coherent diffraction domains, and density of dislocations, it has been proposed that the fraction of powders effectively processed at each collision experiences processing conditions similar to the ones imposed by high-pressure torsion.107
The two mechanical processing methods can be suitably compared only if, in the case of ball milling, attention is focused on the processes occurring at individual collisions. At collision, the powder particles trapped between the surfaces of the colliding milling tools experience a sudden mechanical loading at relatively high strain rates, which gives rise to local mechanical stresses inhomogeneously distributed across the network of contacts between the particles. Under the effect of mechanical forces, the granular bed undergoes a rearrangement characterized by the compression of particles while their surfaces slide against each other, and by the occurrence of severe mechanical deformation.108 Local deformations create the conditions for the activation of mechanochemical transformations, which then occur at individual collisions under processing conditions relatively similar to the ones arising during high-pressure torsion.
However, ball milling is more complicated than high-pressure torsion. Whereas the latter is a continuous mechanical processing method in which, except for intrinsic inhomogeneities, the whole sample experiences the same processing conditions at any given time, ball milling is the typical example for discontinuous methods. The mechanochemical transformation is activated by the mechanical stresses operating at collision, but only a small amount of the powder, randomly selected within the whole powder charge, is involved in each collision. Furthermore, only a small fraction of such amount experiences the necessary conditions to transform.109,110
These features make the powder processing by ball milling an inherently discrete and statistical method. In this regard, it is worth noting that the average duration of collisions, of about 1 ms or less,111 is considerably longer than the time interval underlying the local transformations induced by strain, which roughly occur in 1 to 10 ns.45–59 It follows that a given collision can activate various local deformation processes. Also, any given local process activated by a given collision can reach completion before the successive collision takes place. Under these circumstances, the kinetics of the mechanically activated transformation is governed by the total number of collisions and not by time.112–115
Based on such premises, the kinetics of the mechanochemical process can be described in terms of discrete consecutive stages.112–115 A simplified mathematical model can be developed, which leads to extremely versatile functions that can be used to best fit the experimental data. In turn, this permits information to be obtained on the average amount of powder that experiences at collision the loading conditions necessary to transform, and on the minimum number of effective loading events that the powder must experience before transforming into an intermediate or final product. The practical use of the model is limited by the availability of refined experimental points able to accurately describe the kinetics of the mechanochemical transformations.
3. Phase transformations
It is known that the action of mechanical forces on solid substances frequently leads to polymorphism, i.e. transformation of one crystal structure into another one without chemical change. In this process, the more ordered phases come into existence.91According to thermodynamics, a system at constant pressure and temperature shows a tendency to reach the minimum value of its free energy. If it holds GA > GB for two phases, then phase A is liable to turn into phase B. The relationship between pressure p or temperature T and the volumes of both phases is described by the Clausius-Clapeyron equation
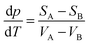 | (4) |
where SA and VA are respectively the entropy and molar volume of phase A. Provided VA > VB, then it holds that dp/dT > 0 (the pressure increases with temperature) and the equilibrium shifts to formation of the phase with smaller volume. If VA < VB then dp/dT < 0 and formation of the phase with larger volume is favoured. The laws of polymorphous transformations of solid substances were described by Buerger in context with coordination of the structure of these substances.116 Later on, the description based on the transformation mechanism was proposed.117
The course of mechanically stimulated phase transformations of some solids was analyzed by Avvakumov.91 He has alleged that the high local pressures and temperatures at the contact surface of the mechanically activated particles as well as the presence of volume defects are responsible for the phase transformations. A peculiarity of mechanically stimulated phase transformations is the formation of phases with higher density, unlike thermal transformations where a phase with lower density usually arises.
3.1 Oxidic compounds
The formation of a non-equilibrium phase by milling of oxidic compounds has been also reported in the literature. PbO118–123 and CaCO3123–132 were taken in the past as reference materials by many authors to study the factors that influence phase transformation by milling. The transformations of litharge → massicot and massicot → litharge were reported for milling of PbO and the transformation aragonite → calcite and calcite → aragonite were observed during the milling of calcium carbonate. The kinetics of the massicot → litharge (massicot = orthorhombic α-PbO, litharge = tetragonal β-PbO) polymorphous transformation in a rotation ball mill has been studied121,133| α-PbO → β-PbO | (5) |
In the case of PbO transformations, the mechanochemical reaction proceeds in two successive stages: (i) the increase of the litharge volume fraction in accordance with a modified logistic growth function, and (ii) the establishment of “mechanochemical equilibrium” at a high transformed volume fraction. A modified logistic growth function of the form
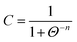 | (6) |
where C is the volume fraction of the transformed new phase at time t, n is the time exponent and Θ = t/tC = 0.5, was found to fit the results of the first stage of transformation up to a certain high value of transformed volume fraction. At this value, the second stage result deviates from the linear plot of ln[C/(1 − C)] vs. lnΘ. The rate dC/dt decreases and tends to dC/dt = 0, where the system stabilizes at a certain mixture value of C. This value is specific to the milling regime. Justification for the use of eqn (6) for the PbO phase transformation is based on the claim that the process is activated by displacement shear.134 This point was confirmed experimentally by high-pressure experiments.119 The authors showed that although massicot is the high-temperature and high-pressure polymorph with higher density than litharge, which is the low-temperature and low-pressure polymorph, both phases may exist, in a metastable form, each in the field of the other equilibrium state. Both will transform into the stable polymorph once replacement shears are produced during milling, which means that a small amount of mechanical action triggers the nucleation of the other polymorph.
It has very often been suggested that pure hydrostatic pressure causes the transformation towards the more dense phase, while the stabilization of the high-temperature phases is due to the local heating generated by the impact of the balls. Suryanarayana24 has reviewed the mechanochemical phase transformation of chalcogenides and has also concluded that the high pressures generated during milling should be sufficient to stabilize the high-pressure polymorph of phases at atmospheric pressure. However, the cyclic calcite → aragonite → calcite (calcite = trigonal CaCO3, aragonite = orthorombic CaCO3) transformations during milling of calcium carbonate reported in previous papers127,128,132a do not seem to be compatible with this interpretation. An alternative explanation would be to assume that the milling creates reticular defects which can store energy greater than the difference of free energy existing between the two polymorphic forms at room temperature. The storage of extra energy in milled materials has been demonstrated in the literature.135–140 Delogu and Cocco9g have criticized that the hot spots generated during milling would be considered the driving force of phase transformations.
The influence of milling on the phase transformation of titania with and without dopants has been widely investigated141–156 because of the industrial interest in this material as a photocatalyst. It has been generally reported that the anatase → rutile transformation takes place through the intermediate formation of a non-equilibrium high-pressure phase. An excellent review on the influence of the structural changes induced by milling of TiO2 on its photocatalytic activity has been recently published by Yin et al.157
A non-equilibrium tetragonal → monoclinic phase transition has been recently reported during the milling of zirconia partially stabilized with ytria.107 On the other hand, it has been observed that the milling of the zirconia gel lead to the stabilization of the metastable tetragonal phase obtained from the thermal decomposition of zirconia gel.158,159
3.2 Sulphides
Zinc sulphide (ZnS) occurs in two main structural forms, i.e. as cubic sphalerite and hexagonal wurtzite. These two minerals represent the terminal members of the series of compounds called polytypes.160 The structure of individual polytypes is variable because of different sequence of layers and different periodicity.161 It is known that such structures contain dislocations, the frequency of which is in many cases dependent on the energy needed for mutual displacement of individual layers.162 The cubic form is thermodynamically stable at low temperature and the hexagonal form is stable at high temperature. The transition of one form to another one can be achieved by high temperature (Fig. 11). The cubic form sphalerite is trasformed into hexagonal form wurtzite at 1020 °C. By slowly cooling, the reverse transformation arises and the cubic form is formed again from the hexagonal form at 850 °C.163 In nature, the cubic form is considerably more common than the hexagonal form, confirming the greater stability of sphalerite at low temperature. The transformation of wurtzite to sphalerite at low temperatures is extremely slow with both phases being present in some mineral assemblages.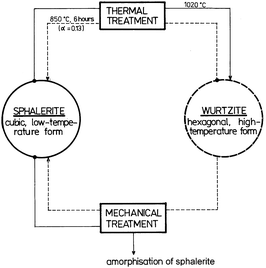 | ||
| Fig. 11 Relations between polymorphs of ZnS. Reprinted with permission from ref. 6. Copyright 2000, Elsevier. | ||
The influence of mechanical activation on phase transformations was carefully studied.91,164–167 It was found that the mechanism of the transformations stimulated by mechanical activation differs from the mechanism induced by thermal treatment (Fig. 11). While the effect of mechanical stress on sphalerite results only in its amorphization, the mechanical activation of wurtzite can bring about transformation into sphalerite (with an associated density increase from 3.48 g cm−3 to 4.09 g cm−3). Senna assumes that the mechanochemical transformation is energetically more profitable than that produced by thermal processing.166 While the thermal transformation requires excitation of all atoms in the structure to an approximately equal degree, only a limited number of nucleation centers is needed to start the mechanochemical transformation. We may assume that the driving force of the mechanochemical transformation of wurtzite to sphalerite is motion of the dislocations in the activated solid phase.91
4. Single-phase nanosystems
Various processing routes have been developed for synthesis of nanoparticles including vapor-, liquid- and solid-state processing routes and combined methods. Here physical vapor deposition, thermal evaporation, sputtering, laser ablation, chemical vapor deposition, spray processing, sol–gel processing, micelles methods and precipitation methods can be mentioned.7 Unlike many of the these methods, the mechanochemical approach produces nanostructures not by cluster assembly but by the structural decomposition of coarse-grained structures as the result of severe plastic deformation. It should be noted that nanoparticles formed under mechanical treatment can be monocrystalline or can be composed of many nanocrystals (coherent scattering regions, CSR) separated by interfaces or amorphous layers. A combination of various physical methods such as HRTEM, analysis of X-ray broadening, measurements of specific surface area, Raman scattering and others is used for comparison of particle and nanocrystalline size. Table 2 presents some literature findings on mechanical activation of several brittle substances.168–176Table 2 Comparison of limiting average particle size and nanocrystalline CSR size for some brittle substances obtained by mechanical activation (MA) and other methods
| Method | Substance | Particle size (nm) | Nanocrystalline CSR size (nm) |
|---|---|---|---|
| MA | h-BN168,169 | 5–10169 | 5–10169 |
| MA | MoO3170 | 60170a,171 | 30171 |
| MA | Graphite172,173 | 1000173 | 20173 |
| MA | Si174,175 | 100175 | 4–40175 |
| MA + etching | Si176 | 45176 | 45176 |
| From nanocomposites | Si176 | 30176 | 30176 |
At the initial stage of MA of brittle substances, the main process is the comminution of particles and formation of new surfaces. A typical curve for increase of specific surface area is shown in Fig. 12. The specific surface area reaches a maximum of 300–450 m2 g−1. The maximum size of nanoparticles for hexagonal boron nitride (h-BN) and for molybdenum oxide (MoO3) was found to be equal to the approximate size of nanocrystalline CSR (Table 2), while the value of specific surface area was found to correspond to the outer surface of the formed nanoparticles. In the case of Si, there is a wide size distribution of nanocrystalline CSR, with the particle size being significantly larger than the size of the CSR. For graphite, a significant aggregation is observed and a particle size is two orders of magnitude larger than the size of crystalline region.
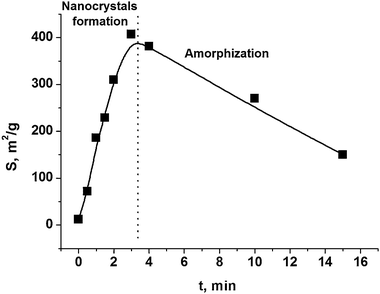 | ||
| Fig. 12 The two stages of mechanical activation of an individual substance, using the example of hexagonal BN. S – specific surface area, t – duration of mechanical activation. Reprinted with permission from ref. 169. Copyright 2009, Elsevier. | ||
The stage of particle comminution is accompanied by the increase in number of point defects and dislocations, and the increase of the lattice parameter and partial amorphization. After the limiting specific surface area is achieved, the matter undergoes amorphization with constant or even decreasing specific surface area. The amorphous phase is mostly located on the particle surface (core/shell structure).177
The presence of a destruction limit in brittle materials is confirmed by Fig. 12. Two approaches are known for the preparation of brittle material nanoparticles with a size smaller than the limiting size.176 Chemical etching of Si after mechanical activation leads to substantial reduction in particle size, which allows practically monocrystalline Si to be obtained (Table 2). A second approach is making a nanocomposite (Si/X) and subsequently dissolving the second component (X).
Numerous studies178–184 of ball milling of elastic metals consider the process of formation of nano-dimensioned states. In that case, the particles are usually not only reduced in size. Their size can even increase, while nanosized regions of crystallinity within large particles are formed as a consequence of plastic deformation. Several mechanisms of nanophase formation are considered: evolution of dislocation structure,179 formation of twins,180etc. It is known that a limiting size of the nanocrystalline regions exists, which depends on the nature of material.181 It is often assumed that the limiting size is determined by the equilibrium between the processes of nanophase formation and re-crystallization.182 There are other explanations to limiting size as well.183,184
Formation of nanoparticles that contain a high concentration of different types of defects leads to significant increase in reactivity of a substance. Under mechanical treatment of graphite185,186 and h-BN187 in gas atmosphere it was possible to chemically absorb up to 2.6 wt% of hydrogen. The amount of Si being oxidized under ball milling can reach up to 20% at room temperature.188 After mechanical activation, h-BN becomes soluble in water.189a The mostly amorphous phase of the surface participates in this reaction and, after long activation times, more than 50% of the material can get dissolved. In fact, the increased reactivity of activated BN is used for growth of nanotubes,189b functionalization of h-BN nanosheets,189c and production of hard cubic BN.168b
4.1 Elements and alloys
Amorphization mechanism. The transformation from crystalline to amorphous phase depends on the ability of the crystalline phase formed by mechanical alloying or mechanical activation to sustain a nanostructured state. In other words, the grain size of systems which can possibly amorphize are expected to be close to a critical germination size. Transformation from a crystalline phase to an amorphous phase was first reported to occur for various elements or alloys with a diamond-type cubic structure such as Si,190 Ge,191 GeSi,192 and GaAs.193
As previously stated,194 mechanical milling (Si, Ge, GaAs) and/or mechanical alloying (Ge–Si) leads to a refinement of the grain size. Two critical grain sizes are found experimentally. The first, d1 (in the range of 20 to 30 nm for ball-milled silicon or germanium), is the grain size below which the lattice expands significantly (up to 0.2% and 0.4%). The second critical grain size, d2 (8 and 4 nm for Si and Ge, respectively), is the grain size below which the transformation from crystalline to amorphous phase takes place (d2 < d1). The size d1 has been proposed to be the size below which the surface energy becomes larger than that of the volume energy. Lattice expansion is a way to compensate this energy difference while keeping the total number of atoms constant (isolated grain hypothesis). Such a mechanism cannot be effective at any size smaller than d2 and a new mechanism, namely the crystal to amorphous phase transformation, must operate below this second critical value. The problem that remains open is that the refinement mechanism allows crystals to decrease so strongly in size. Gaffet and Harmelin190 proposed a two-stage mechanism to explain the Si amorphization under milling: (i) under shock, transformation from Si(I) to Si(II) occurs; (ii) after the shock, when localized pressure release is applied, transformation from Si(II) to Si(III) takes place. When Si(I) is exposed to a shock, Si(II) is formed. This phase is transformed into Si(III) after another shock. When the process is finished, Si(III) is refined. Si(I), Si(II), and Si(III) exhibit different structures, namely the diamond cubic (initial state), α-Sn, and bcc structures, respectively. Such Si(II) and Si(III) structures are indeed observed in high-pressure experiments. The Si(I) → Si(II) phase transition occurs when silicon is compressed during mechanical shocks up to 11.3 GPa, leading to a relative volume decrease of 22%. During pressure release, Si(II) transforms into Si(III) at 7.5 GPa. The Si(III) volume per atom is equal to 92% of that of Si(I). The volume increase during the transformation from Si(II) to Si(III) induces mechanical strains, leading to a breakage of the material and thus to grain refinement. Further works performed in particular by Shen et al. have confirmed the crystalline to amorphous phase transition induced by ball milling.195,196 Two amorphous phases and nanocrystalline Si were produced by ball milling of polycrystalline elemental Si. The nanocrystalline component contains defects such as dislocations, twins, and stacking faults which are typical for coarse-grained polycrystalline materials. The estimated volume fraction of amorphous Si is about 15% while the average size of nanocrystalline grains is about 8 nm. Oxygen-free amorphous elemental Si can be obtained by ball milling. The amorphous Si and the size of nanocrystallites are distributed heterogenously throughout the milled powder. The amorphous Si is concentrated near the surface of milled particles while the grain size of nanocrystalline Si ranges from 3 to 20 nm. Two possible amorphization mechanisms (i.e. pressure-induced amorphization and crystallite refinement-induced amorphization) are proposed for the amorphization of Si induced by ball milling. Such an amorphization of silicon induced by mechanical treatment has also been investigated by Streletskii et al.197,198 The mechanical treatment of silicon in an inert atmosphere is accompanied by the formation of two populations (coarse and fine) of nanoparticles along with the formation of an amorphous phase. It may be concluded that the decrease of grain size below the critical value of about 4 nm is accompanied by the amorphization of silicon. The amount of amorphous phase for the maximum injected energy (500 kJ g−1) exceeds 40%. Particular attention has been paid to the cathodoluminescence of mechanically milled silicon.199 Nanocrystalline silicon has been produced by ball milling of silicon single crystals. The milled powder shows cathodoluminescence in the visible and near-infrared ranges. The main visible band appears at about 1.61–1.64 eV and is attributed to the presence of nanocrystals. An intense infrared band, which is not systematically observed in nanocrystalline silicon structures, appears in milled silicon at the same energy (0.8 eV) as the well-known dislocation luminescence band of silicon. It is suggested that this emission arises from the high density of extended defects generated during the milling treatment.
Švrček et al.200 have presented a different approach to fabricate very fine silicon nanocrystals with quantum confinement size ex situ from the silicon dioxide matrix, namely by ball milling and pulverizing of porous silicon. The authors have shown that light-scattering experiments can reveal some additional properties of Si nanocrystals dispersed in liquid solutions: introducing ammonia into the ethanol colloidal solution containing Si nanocrystals leads to temporal declustering of both types of Si nanocrystals. The particle sizes calculated from the scattering experiments are in good agreement with those obtained from HRTEM analysis. Si nanocrystals are in high concentration in the samples fabricated by pulverizing of porous silicon and are embedded into spin-on glasses (pure spin-on glass or its phosphor-doped solution) and show bright visible photoluminiscence (PL) due to their size quantum effect and surface states. The Si nanocrystals fabricated by ball milling are embedded into amorphous tissue containing defects. Amorphous tissue is a considerable source for non-radiative recombination processes. Si clusters prepared in the form of self-supporting samples embedded in a phosphor-doped spin-on glass matrix exhibit weak PL at room temperature with maxima around 700 nm.
Experiments show that mechanical attrition can induce complete crystallization of an amorphous solid and complete amorphization of a nanocrystalline phase, as shown in the case of Se.201 The induced phase transformation has been found to be closely related to the initial microstructure of the processed materials. Depending on the starting materials, mechanical attrition may either (i) raise the excess energy of the milled sample by accumulating various microdefects or (ii) lower the total excess energy by activating a phase transformation to a more stable state (such as crystallization). Some calorimetric experiments202 have shown that during cryogenic temperature milling, amorphization is faster than at ambient temperature. The experimental results have been interpreted as a crystalline to amorphous phase transition driven by defects created by frequent mechanical deformation.
X-ray diffraction (XRD) investigations and thermal analyses have revealed that the amorphization onset corresponds to a critical size and a decrease in microstrain.203 During the major amorphization process, the remaining crystallite size remains unchanged with a constant lattice expansion. The amorphization kinetics was explained in terms of crystallite destabilization model rather than by the classical shell kinetic model. The interpretation of the latter model has been questioned by Caro and Van Swygenhoven working in the field of computer simulations.204 For these amorphous Se-annealed samples, a correct calculation of grain boundary area and energy balance in the nanocrystalline phase yields a grain boundary width of 2.5 nm and a grain boundary energy of 0.4 J m−2. The grain boundary width is significantly larger than in simulated nanocrystalline nickel. An incorrect description that does not take into account the finite width of the grain boundary leads thus to an apparent decrease of the grain boundary energy with decreasing grain size. Nasu et al.205 analyzed the structure of amorphous Ge–Se prepared by mechanical milling. The amorphization process and the short-range order structure of amorphous GeSe2, Ge3Se4, and Ge4Se5 prepared by mechanical milling was investigated. GeSe2 becomes amorphous over a shorter milling time. Much longer time is needed to amorphize Ge-enriched alloys such as Ge4Se5. Amorphous GeSe2 and Ge3Se2 prepared by mechanical milling exhibit a local order similar to the corresponding amorphous phases prepared by liquid quenching, that is the covalent 4(Ge)–2(Se) folded structure. The amorphous Ge4Se5 (which cannot be obtained by liquid quenching) also has the 4–2 folded structure, although its composition is nearer to GeSe. Such a crystalline–amorphous transition of Ge–Se alloys by mechanical milling has also been investigated by Tani et al.206 The mechanisms for amorphization of Se are suggested to be based on the distortion between helical chains connected by van der Waals forces. Monoclinic GeSe2 is amorphized by disordering of the bonds between the connected tetrahedra. The tetrahedral unit of GeSe2 is greatly distorted by mechanical activation. GeSe2 is completely amorphized after 4 hours of milling, and Se after 30 hours under the given milling conditions. This difference has been considered an indication that disordering between the connected tetrahedral units in monoclinic GeSe2 is induced more easily than it is between chains in trigonal Se.
Graphite. Huang investigated the structure of amorphous-like graphite and defects induced by ball milling by HRTEM and electron energy loss spectroscopy.207 The deformed structures and the amorphization kinetics were clearly revealed by these methods. Basal plane stacking disorder, cleavage, delamination cracks, misorientation bands and low angle (0002) twist boundaries are observed. The basal plane stacking disorder is probably produced by a simultaneous shearing. Other defects such as number of half Frank loops or interstitial loops, and bending and buckling of the basal planes are also observed. Frequent shearing of (0002) planes acting in parallel with the increase of the Frank loop density finally leads to the breakage of the hexagonal network at a very fine scale and an amorphous-like structure is formed. The amorphous-like phase comprises highly curled flakes and it exhibits sp2 hybridization. The amorphization kinetics has been found to correspond to a defect-controlled process. Welham and Williams have also studied extended milling of graphite and activated carbon.208 Graphite and activated carbon were ball-milled under vacuum for 1000 hours. Graphite became amorphous, with no evidence of recrystallization after heating up to 1200 °C. Little change in the structure of the activated carbon was observed. Heating two milled samples under argon showed that they absorbed substantial gas amounts – over 30% of their mass in some cases. The temperature of oxidation of graphite and activated carbon is decreased in the case of longer milling times. The greatest decrease in ignition temperature for graphite was concomitant with the largest increase in noncrystalline carbon, indicating the role of disordered carbon in enhancing the oxidation. Indeed, graphite milled for 1000 hours showed reactivity similar to that of activated carbon. The structural disorder and phase transformation in graphite induced by ball milling was also investigated by Shen et al.209 It was shown that nanocrystalline graphite with a crystallite size of about 2 nm is formed after 8 hours of ball milling. Further milling produces a mixture of nanocrystalline and amorphous phases. Relatively large structural disorder is induced in the ball-milled graphite, as revealed by XRD and Raman scattering experiments. Furthermore, the nanocrystalline graphite is relatively stable when compared with some nanocrystalline metals. Salver-Disma et al. performed TEM studies of carbon materials prepared by mechanical milling.210 Mechanical milling has been applied to generate an increasing amount of disordered carbon at a rate depending on the type of milling mode used (shear- or shock-type modes). When the shock-type milling is used, the triperiodic structure and the lamellar microtexture of graphite completely breaks down to give microporous and turbostratic carbons made of misoriented basic structural units. Graphite milling permits the elaboration of disordered carbons. The involved mechanism differs from a simple reverse graphitization, since not only the structure but also the microtexture is strongly modified by milling. After heat treatment at 2800 °C, the graphite structure is not formed again and a mesoporous turbostatic carbon is mainly obtained. Shear milling has been found to be less effective since remnants of graphitic carbon are still present within the disordered carbon after the process. In order to achieve lithium intercalation, a milling process has been performed in graphite and soft carbons.211,212 The effects of milling on the morphology and electrochemical performance of graphite and soft carbon powders with respect to lithium insertion were studied. The morphology of the milled graphite powders was found to depend strongly upon the nature of the interactions (i.e. impact or shear- or shock-type modes) generated by two kinds of mixer mills used. For the same milling time, crystallite size was the smallest and the density of defects the highest for graphitic powders that were ball-milled using impact interactions. For each milling mode, the resulting ball-milled carbonaceous powders were found to be totally independent from the nature of the precursor used (graphite, carbon, coke) and/or from its morphology (layers, microbeads and fibers). The effect of liquid media during milling of graphite has been investigated by Janot and Guerard.213,214 Ball milling of graphite in the presence of liquid leads to very thin, well-crystallized, and anisometric particles. In the presence of water, the graphite particles are covered by γ-Fe2O3 (maghemite), which is formed by the oxidation of the ferrous alloy coming from the milling tools. In the presence of n-dodecane, well-crystallized and highly anisotropic particles of graphite exhibiting promising features as anodic materials for lithium-ion batteries can be synthesized.
Diamond. A new fabrication method based on wet milling to produce homogeneously fluorescent nanodiamonds with high yields is described in ref. 214b. The powder obtained by high-energy ball milling of fluorescent high-pressure high-temperature diamond microcrystals was converted into a pure highly concentrated aqueous colloidal suspension of highly crystalline ultrasmall nanoparticles with a mean size less than or equal to 10 nm. The whole fabrication yield of colloidal quasi-spherical nanodiamonds was several orders of magnitude higher than those previously reported starting from microdiamonds. The results open up avenues for the industrial cost-effective production of fluorescent nanodiamonds with well-controlled properties.214b
4.2 Oxides
Polymorphic transformation and amorphization of oxides under mechanical stressing have been known for the long time and are well documented.215–217 Particularly, these phenomena on downsizing to a nanometric regime has recently been intensively studied, including their atomic figures in conjunction with well-defined high-pressure systems.218,219 Information acquired in the aforementioned studies is closely related with crystallization of oxides from their amorphous precursors, which is only completed when the rest of the impurity species including OH are eliminated. These points were studied for TiO2 and ZrO2 inorganic gels in detail.220,221 A vast number of studies on the direct synthesis of complex oxides, associated with their electronic application, have been carried out.222 Detailed structural analyses enabled elucidation of the mechanochemical reaction processes.223,224 One of the most important features of these processes is the simplicity of a one-step mechanism. When starting materials contain precursors of the component oxides, e.g. carbonates, an intermediate-stage-like carbonate complex was obtained.224 High-resolution TEM reveals that the mechanosynthesized products are well crystallized in the core part, with a disordered near-surface shell region.223The preparation of nanocrystalline complex oxides based on mechanochemical synthesis (MCS) is a rapidly expanding area of solid-state science.7,225–241 In this sense MCS represents a comparatively new technological approach to preparation of ultra-disperse nanosized oxides without any thermal treatment (one-step mechanochemical route)225,230 or with some partial heating at a lower temperature and for a shorter time.233 Distinctive features of the complex oxides prepared by the MCS method are the new compounds235,242 or the new energetic,225 structural,227 dispersion227 or electronic states of already known compositions.243 The mechanochemical processing provides an opportunity to fabricate novel nanomaterials with anomalous properties different from those of bulk samples prepared by conventional (ceramic, sintering, co-precipitation, hydrothermal) processing.7,241 The explanations of these effects are related to some experimentally established facts. Thus, MCS initiates changes of particle size,227 appearance of defects (structural disorder, lattice strains, excitation and migrations of defects, formation of vacancies),225,229,242,244 structural transformations (polymorphous conversions, partial or complete amorphization, changes of coordination state of ion)225,227 and changes in the phase composition.225,229 The steps in the MCS procedure are interconnected elastic, plastic, and electronic deformations, mechanochemical decomposition, and synthesis.229 The effect of MCS of complex oxides depends on the following factors:
(i) Type and intensity of the impact, nature of the medium and the conditions under which this effect is applied, i.e. the choice of the method and selection of the conditions of mechanical treatment.227,232,245 Vibration mills have been applied rarely,236 while planetary ball mills are often used.225–232 In the latter case, the mechanical treatment involving velocities of the rotating body up to 100, 250, and 390 rpm is considered to be low-energy, medium-energy and high-energy processing, respectively.245 The ratios of the weight of a ball in a ball mill to the weight of the sample being ground (BPR) that have been applied were as follows: 50:1,225 30:1,241d 20:1,226,227,237,242,243 15:1,233 13:1230 and 10:1.231 The absence of normative regulations with respect to the conditions of treatment does not allow results to be obtained on the same basis that could be compared regarding the mechanochemical activation of powders having one and the same chemical nature.
(ii) The rate of scattering (dissipation) of the transferred energy, i.e. depending on the chemical nature of the composite upon which the impact is focused. Using MCS, different types of complex oxides have been prepared successfully: ferrites MFe2O4 (M = Ni2+, Cu2+, Co2+, Mg2+);225,236,243 hexaferrites Ba(M)2xFe12−2xO19 (M = Ni2+, Zn2+, Sn2+);235 perovskites and perovskite-like compounds;241 complex oxides with fluorite Bi1.6M0.4O3 (M = La, Y, Yn, Ca) and pyrochlore Gd2Ti2O7, Gd2(Sn1–xZrx)2O7 crystal structure;241 garnets Y3Fe5O12, SmxBiY2–xFe5O12;238 oxide sheelite PbMoO4;240 solid solutions;235,243etc.
The metastable character of mechanochemically synthesized complex oxides is distinctly manifested especially in the case of nanosized powders. This is displayed as local non-homogeneities or as an evolution of the composition, sometimes decomposing spontaneously into new nanocomposite materials of various oxide composition. The aggregation of mixed oxide powders in the course of the mechanochemical procedure is inevitable. Moreover, there exists a clearly expressed correlation: the faster the mechanochemical synthesis of complex oxides proceeds, the more intensive is the process of aggregation.241 In nanostructured materials, the high surface area makes surface effects become of major importance. Recently, it was observed that ball milling of ferrite samples can also lead to changes in the metal site population.244 Owing to the thermally induced variability of the cation arrangement and the spin configuration of ground ferrite materials annealed at different temperatures, a wide range of magnetic behaviour can be observed.225,227,228,244
Mechanochemically synthesised hexaferrites and substituted hexaferrites are useful materials for various applications. ZnRu- and NiRu-substituted hexaferrite powders Ba(M)2xFe12–2xO19 rapidly diminished the coercivity to suitable values for high-density magnetic recording applications. Alternatively, NiSn, SnRu, ZnRu, and NiRu mixtures showed a potential for application as materials for microwave absorption.235
The complex oxides of perovskite structure, possessing high electron conductivity and mixed type of conductivity, represent a huge group of compounds with various structures.239 Depending on their function as materials, different precursors are used for the mechanochemical synthesis: a direct route from oxides or their hydrated forms, or an indirect route from salts, including use of diluents. This is associated with their wide application in fuel cells, electrochemical devices, catalysts or conducting membranes. The mechanochemical synthesis at room temperature is accomplished within time intervals of about 30 minutes, whatever the complexity of their composition might be. The necessary duration of the process depends on the melting point of the oxides, initial dispersion, and degree of oxidation.7,241
The evolutionary changes in composition, the initial materials, the intermediate products, and the final complex oxide products are characterized by many methods. These include XRD (for determination of phase composition, structural parameters, average effective crystallite sizes, and levels of microtensions),225–228,230–235,242–245 Mössbauer spectroscopy (phase composition and magnetic structure),228,231,232,234,235,237,242–245 IR spectroscopy (identification, phase-structural changes),241d thermal analysis (thermal behaviour and phase changes),225,226,241d XPS (electronic structure and surface composition),233 magnetic measurements (magnetisation experiments),231,235,242,243 electron microscopy (particle sizes, particle aggregation),228,230,231,233,236 Raman spectroscopy,228 temperature-programmed reduction (TPR),237 AFM,233etc.
4.3 Other oxidic systems
Complex metal oxides with the perovskite structure and their composition have attracted the interest of chemists for a long time as they exhibit interesting solid-state properties making the structures very appropriate for application as catalysts, electroceramics and others.246,247 Well-known perovskites are compounds based on lead (PbTiO3, PbMg1−xNbxO3, Pb1−xSrxTiO3 and Pb1−xLaxZr1−xTixO3). These compounds are not environmentally friendly because of the volatility and toxicity of PbO. Because of unique dielectric, piezoelectric, thermoelectric, optical and ferroelectric properties of titanates of alkaline earth metals, they have become an object of study of many research groups. There are different methods of their synthesis,248–251e.g. high-temperature solid-state techniques, hydrothermal processes, sol–gel processes, co-precipitation methods, as well as mechanochemical treatment – an alternative method which complies with green chemistry. Mechanochemical methods offer simplicity, one-step operation, solvent-free synthesis and/or modification of properties of advanced products.252,253The ternary oxide system CaO–TiO2–CuO is a very interesting material for application as capacitors. This material was investigated in detail for CaTiO3 and its modification by substitution of calcium (Ca1−xCuxTiO3, 0 ≤ x ≤ 0.75) and titanium sub-lattice (CaTi1−xCuxO3, 0 ≤ x ≤ 0.4) by copper.254–260
The influence of mechanochemical treatment on the properties of the resulting ceramics was also determined, especially taking into account the ball material (steel, ZrO2). Ca0.25Cu0.75TiO3 ceramics (CCTO) has been chosen as attractive material for microelectronic devices because of a high value of dielectric permittivity (ε′).261–263 There is an exemplary illustration of the dielectric properties of Ca0.25Cu0.75TiO3 with doped metallic iron (CCTO/Fe), zirconia (CCTO/Zr) and, for comparison, CCTO prepared by high-temperature treatment (CCTO/T) below.263–266Fig. 13a and b show dielectric properties of studied perovskite-related CCTO compounds as a temperature relationship of, respectively, the real component of dielectric permittivity (ε′) and dielectric loss (tan δ) at a frequency of 1 kHz. The presence of a small amount of zirconia (CCTO/Zr) causes the smallest changes in the electric permittivity (ε′) with increasing temperature, similar to the nature of CCTO/T. From the practical point of view such an effect as well as very low and stable value of dielectric loss (tan δ) in the temperature range of −50 to 50 °C is very convenient. Material with the presence of metallic iron (CCTO/Fe) is different from the CCTO/Zr and CCTO/T ones. From the ambient temperature, a significant increase in the value of ε′ is observed. At 200 °C, ε′ reaches a value of about 3000. However, high values of dielectric loss disqualify such ceramics for practical applications as capacitor materials, indicating the possibility of material overheating. It can be stated that mechanochemical synthesis is useful for production and modification of Ca0.25Cu0.75TiO3 (CCTO). In comparison with ceramics, CCTO/T formed by high-temperature treatment, the presence of ZrO2 (CCTO/Zr) improves Ca0.25Cu0.75TiO3 functional properties in terms of ceramic capacitors.
 | ||
| Fig. 13 Temperature relationship (a) of the real component of dielectric permittivity (ε′) and (b) dielectric loss (tan δ) at a frequency of 1 kHz for Ca0.25Cu0.75TiO3 (CCTO) mechanochemically synthesized under different conditions: ■ CCTO/Fe – CCTO with the presence of metallic iron; ○ CCTO/T – CCTO by high-temperature treatment; ▲ CCTO/Zr – CCTO with the presence of zirconia.261 | ||
The mechanism of the impact through applying mechanical activation is based on the understanding that the mechanical energy is almost entirely transformed into thermal energy, whereupon some physical and chemical processes are initiated. In this sense, the MCS of complex oxides can be regarded as a process of “quasi-adiabatic” accumulation and distribution of energy in a solid-state body as a consequence of the mechanical treatment, possessing sufficient intensity of the impulse impact in a small volume for a very short time interval.
The transferred energy is dissipated in the mechanically treated solids via different mechanisms depending on its chemical nature. In the case of metals, the thermal conductivity is basically determined by the movement of the electrons, i.e. an electronic mechanism, while in the case of semiconductors the transfer of energy is accomplished in a mixed form: phonon and electron mechanisms. In the case of non-conducting crystals, which happens to be the case of most complex oxides, the mechanism of thermal conductivity is entirely based on phonons. The average free path of the phonon is inversely proportional to the phonon energy and, accordingly, inversely proportional to the temperature. The temperature dependence of the mean free path of the phonons in the metal oxides has a complicated character.267 The thermal conductivity, accomplished by means of a phonon gas, strongly depends on the mechanism of dissipation of the phonons and on their joint interaction with one another and with the defects of the crystal lattice. In the case of phonon–phonon interaction, there appears a new phonon with an impulse different from the resultant impulse of the two interacting phonons and, in this case, there is also a change in the direction of the stream of energy. At high temperatures (above Debye’s temperature), the mean free path is proportional to T−1. The scattering of the phonons can occur also from various imperfections of the crystal: defects, admixtures, grain boundaries, near-range disorder and distant-range disorder. Therefore, the above-listed possibilities of energy absorption and quasi-impulse of the phonon will play an important role in MCS of mixed oxides, where the formation of defects in the treated material and its disintegration into smaller particles will always accompany the treatment.
Mechanochemical synthesis has developed into an essential tool for the synthesis of many technologically important ferroelectric oxide systems, including BaTiO3, Pb(Zr,Ti)O3 (PZT) and many others. Extensive reviews on the mechanochemical synthesis of complex ceramic oxides can be found in Kong et al.268 and Rojac and Kosec.269 As already pointed out in the initial part of this section, research and development of piezoelectric materials, which are widely used in applications such as sensors, actuators and transducers,270 have been strongly affected by recent regulations of the European Union, which aim at replacing the toxic lead compounds with environmentally safer materials. This led to intensive research activity, focused primarily on finding a lead-free substitute for the most commercially used piezoelectric material, i.e. PZT.271,272
So far, the (K,Na)NbO3 (KNN) solid solution has been one of the most studied among the alternatives.273 The research on KNN was reinforced in 2004 by the paper of Saito et al.,274 who showed that piezoelectric properties comparable to PZT could be achieved by modifying KNN with Li, Ta and Sb, or by grain orientation (texturing). However, the high piezoelectricity of these materials originates from the enhanced polarizability associated with the compositional shift of the orthorhombic-to-tetragonal polymorphic phase transition (PPT) from 195 °C for pure KNN275 downward to room temperature.271,272 This inevitably leads to temperature-dependent properties and partial depoling of the ceramics by thermal cycling through the PPT.271,276 The interest in KNN has been recently revived by the discovery of the excellent functional properties of the niobate sintered in reducing atmosphere.277 This opens a possibility to process KNN in, for example, multilayer piezoelectric actuators, using inexpensive base-metal electrodes, such as Ni and Cu, instead of the often used noble-metal electrodes.
The solid-state synthesis of KNN, particularly KNN-modified compositions, is challenging. Several issues have been discussed in the literature: (i) high hygroscopicity of the starting alkaline carbonate powders, especially K2CO3, and high volatility of alkaline oxides at elevated temperatures, both creating difficulties in controlling the stoichiometry,275,278–284 (ii) formation of secondary phases, some of which are hygroscopic,275,285 and (iii) difficulty in achieving high compositional homogeneity, especially at the B-site of the perovskite ABO3 lattice, when KNN is modified with, for example, Ta, such as in the case of (K,Na,Li)(Nb,Ta)O3 (KNLNT) solid solution.286,287
KNN is usually prepared by the solid-state reaction, mostly from a mixture of alkaline carbonates and Nb2O5.288 Despite the high technological importance of KNN, remarkable progress in the understanding of the solid-state synthesis of this material has only been made recently. A diffusion couples study showed that the reaction between Na2CO3 and Nb2O5 proceeds by decomposition of the carbonate at the interface and subsequent diffusion of Na+ and O2− ions into the interior of the Nb2O5.289 According to the values of the diffusion coefficients from the literature, the diffusivity of sodium (D600°C 10−12 m2 s−1)290 is considerably higher than that of niobium (D600°C 10−28 m2 s−1),291a confirming the observed diffusion path. Since oxygen diffusion in complex oxides is generally faster than cation diffusion, the overall growth rate of the reaction layer is then controlled by the slower Na+ ions.289
Similar diffusion mechanisms, as proposed for Na2CO3–Nb2O5, were confirmed in the K2CO3–Nb2O5 and in the ternary (Na2CO3 + K2CO3)–Nb2O5 couples.289 However, in systems with K2CO3 the rate constant of the reaction-layer growth at 600 °C was found to be an order of magnitude lower (3 × 10−15 m2 s−1) than in the Na2CO3–Nb2O5 system (10−14 m2 s−1). The growth rate of the reaction layer in the K2CO3-containing systems is thus controlled by the slower K+ ions, which is in agreement with the lower self-diffusion coefficient of K+ (D600°C 10−13 m2 s−1) in alkali-silicate glasses as compared to Na+ (D600°C 10−12 m2 s−1).290
The determined reaction mechanism in the mixtures of alkaline carbonates and Nb2O5 leads to several important consequences associated with the processing of KNN and related materials:
(i) Since the reaction is diffusion-controlled, the temperature range of the carbonate decomposition becomes strongly dependent upon the particle size, especially that of Nb2O5, which determines the diffusion distance.292 Thus, to drive the reaction to completion at a given temperature, extreme care is required in terms of controlling the particle size and size distribution of the starting powders.
(ii) Different diffusion rates of Na+ and K+ may lead to local gradients of concentration.289 To overcome this problem, repeating calcinations with intermediate milling steps and/or increasing temperature and time of annealing may be required.
Further problems arise when Li and Ta are introduced into KNN. The addition of Li2CO3 to the Na2CO3–K2CO3 mixture progressively decreases the eutectic temperature from 710 °C (eutectic molar composition Na2CO3/K2CO3 = 0.58/0.42)293 to 397 °C, which is the temperature of the ternary eutectic with the molar composition Na2CO3/K2CO3/Li2CO3 = 0.3/0.26/0.44.294 Therefore, improper mixing of initial alkaline carbonates with Nb2O5 may lead to local eutectic melting at relatively low temperatures, i.e. before the carbonate decomposition is completed. This may cause de-mixing effects by liquid segregation. The addition of Ta to KNN further complicates the synthesis. The major problem in this case is to achieve high compositional homogeneity at the B-sites of the perovskite due to the very low diffusivities of niobium (D600°C 10−28 m2 s−1)291a and even lower of tantalum (D600°C 10−33 m2 s−1),291b which means almost non-existent interdiffusion at usual calcination temperatures. In fact, studies on KNLNT showed that such inhomogeneities, once formed, were not eliminated by prolonged high-temperature annealing and/or intensive attrition milling applied after each calcination step.286
The key question is whether it is in principle possible to modify the diffusion-limited process to such an extent that it would result in another reaction pathway. In the following we show that this may be achieved by the mechanochemical approach. The mechanochemically assisted synthesis of KNLNT with the composition (K0.485Na0.485Li0.03)(Nb0.8Ta0.2)O3 was performed from Na2CO3–K2CO3–Li2C2O4–Nb2O5–Ta2O5 mixture. Experimental details of the high-energy milling procedure are reported in ref. 295 and 296.
Infrared spectroscopy analysis was used to follow the vibrations of the carbonate ions in the powder mixture during mechanochemical activation. The aim was to obtain insight into possible symmetry changes of the carbonate species. The IR spectra of the starting mixture after different activation periods are shown in Fig. 14a.
 | ||
| Fig. 14 (a) Infrared spectra of Na2CO3–K2CO3–Li2C2O4–Nb2O5–Ta2O5 mixture subjected to mechanochemical activation for different periods. The spectrum of the mixture prepared by a separate activation of the starting powders followed by mixing in acetone is given for comparison (sep. act. + mixing). ν1, ν3 and ν4 denote the vibrations of the CO32− ion. (b) X-ray diffraction (XRD) patterns of the mixture activated for different periods. (c) Normalized areas of Nb2O5 and Ta2O5 peaks from (b) as a function of activation time. Reprinted with permission from ref. 296. Copyright 2010, Wiley. | ||
The most intensive absorption band at 1420 cm−1, appearing in the initial non-activated mixture (Fig. 14a, 0 h), belongs to the asymmetrical C–O stretching vibration of the CO32− ions (ν3) characteristic for both Na2CO3297 and K2CO3.298 Upon milling, considerable changes were observed in the IR spectra, finally resulting in the splitting of the ν3 and activation of the ν1 vibration of the CO32− ion (Fig. 14a, 20 h). These IR spectroscopic changes have been well documented in the literature for various carbonato complexes and were associated with the lowering of the point-group symmetry of the CO32− ion from D3h, which is characteristic for the free, non-coordinated, CO32− ion, to C2v or Cs, which are characteristic for coordinated CO32− groups.296,299–304 The lowering of the symmetry was related to the mechanochemically driven coordination of the CO32− ions to the transition-metal cations (Nb5+, Ta5+). In principle, such a symmetry change can be viewed as a new bond between the CO32− and Nb5+/Ta5+, which effectively distorts the carbonate ion from its original D3h symmetry and consequently leads to the amorphization of Na2CO3 and K2CO3.296 This intermediate phase formed during milling was referred to as the carbonato complex and was identified in various other alkaline-carbonate (Li, Na, K, Rb)–transition-metal oxide (V, Nb, Ta) mixtures.301–305 Note that no symmetry changes were observed if the reagents were activated individually with subsequent mixing in acetone, i.e. the ν3 vibration, shown in Fig. 14a, exhibits essentially no splitting. Thus, the formation of the carbonato complex clearly requires the mechanochemical interaction between the CO32− and the transition-metal oxides.301 Finally, we note that Li2C2O4 apparently affects the kinetics of the formation of the complex (not shown), however its role requires further study.296
In addition to the symmetry change of the CO32−, structural changes of the reaction counterparts, i.e. Nb2O5 and Ta2O5, were observed as well. The X-ray diffraction (XRD) peak intensities of Nb2O5 and Ta2O5 decrease with activation time (Fig. 14b and c), suggesting progressive amorphization of the oxides as the complex is being formed. The tendency of Nb2O5 and Ta2O5 to react with the CO32−, forming the carbonato complex, was further confirmed in binary A2CO3–M2O5 (A = Na, K; M = Nb, Ta) systems.301–303,305 In addition, direct observations of the bonding between Na+ or K+ and Nb5+via the CO32− group were obtained by means of Raman spectroscopy and nuclear magnetic resonance (NMR).224,302 Finally, HRTEM analysis of the activated powder (Fig. 15) revealed a major amorphous phase, embedding small nanocrystallites (10 nm) of either Nb2O5 or Ta2O5.296 The amorphous phase was identified as the carbonato complex, however the presence of the nanocrystallites suggests incomplete bonding of both transition-metal cations to CO32−.
 | ||
| Fig. 15 HRTEM image of the Na2CO3–K2CO3–Li2C2O4–Nb2O5–Ta2O5 powder mixture after 20 h of mechanochemical activation. According to energy-dispersive X-ray spectroscopy (EDXS) analysis, the nanocrystalline particles were identified as either Nb2O5 or Ta2O5, whereas the amorphous phase, i.e. the carbonato complex, consisted of K, Na, Nb and Ta. Note that Li cannot be detected by EDXS analysis. Reprinted with permission from ref. 296. Copyright 2010, Wiley. | ||
In order to evaluate the role of the carbonato complex in the synthesis of the solid solution, we compared the KNLNT ceramics prepared in two ways (Fig. 16): (i) by conventional solid-state synthesis306 and (ii) by mechanochemical activation, followed by a single calcination step.295,296 Both calcined powders were sintered at 1080 °C for 2 h. In the KNLNT ceramics prepared by the conventional route Ta inhomogeneities, shown as bright and dark regions on the SEM micrograph in Fig. 16a, were observed. In contrast, a homogenous solid solution was obtained using the mechanochemically-assisted synthesis (Fig. 16b).288,295 The difference between the two samples was reflected also in the compositional analysis. Much larger fluctuations in Nb and Ta concentrations were found in the conventionally prepared KNLNT than in the mechanochemically activated one (compare Fig. 16c with d).
 | ||
| Fig. 16 Comparison of compositional homogeneity of KNLNT ceramics prepared by conventional (a and c) and mechanochemical routes (b,295 d288). The figures show SEM backscattered-electron (BE) images of the polished surface of the ceramics (a and b) and molar fractions of Na, K, Nb and Ta, determined by EDXS analysis, at randomly selected spots on the surface (c and d). For comparison, an average composition, determined on a 0.3 × 0.3 mm2 surface area, is added. The marked bright and dark regions in (a) correspond to Ta-rich and Ta-poor regions, respectively. Fig. 16b reprinted with permission from ref. 295. Copyright 2008, Wiley. | ||
Earlier studies suggested that the homogeneity of the pre-calcined mixture, particularly the distribution of the low-reactive Ta2O5, determines the final compositional homogeneity of the KNLNT ceramics.286 This is in agreement with the present results (Fig. 16). With respect to Ta2O5, we believe that the origin of the high level of chemical homogeneity of our activated powder is twofold: (i) it is directly attributed to the presence of the carbonato complex, based on the fact that Ta is partially bound to CO32−, and (ii) the unreacted Ta2O5 is present in the form of small nanocrystallites, which are uniformly distributed in the amorphous matrix (Fig. 15), providing short diffusion paths upon heating. It should be noted that the formation of KNLNT upon heating occurs through the decomposition of the carbonato complex, presumably with rather short diffusion distances, which probably allows the problem of the low diffusion rates of the B-site Ta and Nb cations to be overcome. Therefore, the presence of both the carbonato complex and the nanocrystallites contributes to an overall homogeneous distribution of Ta2O5 in the powder, resulting finally in the superior B-site homogeneity of the KNLNT ceramics (Fig. 16b and d).296
In the next part, we show the influence of the observed inhomogeneities in the KNLNT ceramics on their dielectric and piezoelectric properties. Fig. 17 shows the dielectric permittivity as a function of temperature for both conventionally and mechanochemically processed KNLNT. In comparison to the dielectric response of the conventionally processed KNLNT (Fig. 17a), a sharper peak in the permittivity around the Curie temperature (TC = 310 °C) and a higher peak value of the permittivity of the mechanochemically prepared KNLNT (Fig. 17b) support its higher compositional homogeneity. Secondly, the temperature of the O–T phase transition (TO–T) in the conventionally processed KNLNT appears at 50 °C (inset of Fig. 17a), whereas in the mechanochemically processed KNLNT the transition is likely to occur below room temperature (inset of Fig. 17b). This is supported by the XRD analysis (not shown): at room temperature the conventionally prepared KNLNT is orthorhombic,306 whereas the mechanochemically prepared KNLNT could be indexed with the tetragonal structure,295 in agreement with the original paper by Saito et al.274 The observed different TO–T (50 °C) in the conventionally processed KNLNT is probably related to the Ta inhomogeneities, e.g. incomplete incorporation of Ta into the solid solution and presence of Ta-poor(rich) regions with different TO–T (see Fig. 16a).286 Additionally, it could also be related to Li,307 which is, however, difficult to analyze because of its low concentration (3 at% on A-site).
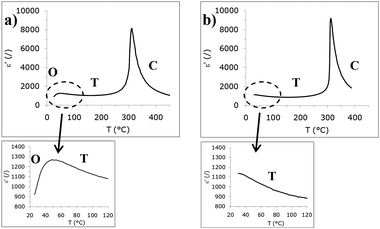 | ||
| Fig. 17 Dielectric permittivity as a function of temperature, measured at 10 kHz, for KNLNT ceramics prepared by (a) conventional and (b) mechanochemical routes. Orthorhombic, tetragonal and cubic phase regions are denoted with O, T and C, respectively. Note the different temperatures of the O–T phase transition of KNLNT ceramics processed by the two routes (insets). | ||
The enhancement of the piezoelectric properties of KNN via the chemical modification is strongly dependent upon the TO–T.271 Being closer to room temperature (see inset of Fig. 17b) and thus providing larger structural instability, the mechanochemically synthesized KNLNT indeed exhibited a 44% higher piezoelectric d33 coefficient (230 pC N−1)295 than the conventionally prepared KNLNT (160 pC N−1). Even if the polymorphic phase boundaries are, in general, less attractive in view of a temperature-stable piezoelectric response, the present work clearly demonstrates the potential of the mechanochemical approach.
Mechanochemically-assisted synthesis enables a simple one-step calcination route for the preparation of highly homogeneous KNLNT ceramics. In contrast to the solid-state reaction mechanism, which is governed by diffusion processes, the mechanochemical activation is characterized by the formation of an amorphous carbonato complex. This phase contributes to the enhanced chemical homogeneity of the resulting powder and allows most of the limitations associated with the diffusion-controlled solid-state synthesis to be overcome. The dielectric and piezoelectric properties of the mechanochemically processed KNLNT are superior compared to those obtained by conventional ceramic processing.
4.4 Sulphides
Sulphides exhibit a great variety of physical, chemical and physico-chemical properties. They display similar structural defects to oxides with cation vacancies, interstitial cations or anionic defects. However, the differences in concentration, structure and mobility of these defects are much more varied in the case of sulphides.308 Sulphides play an important role in traditional technological applications as well as advanced materials. During the last few years, the synthesis and characterization of new transition metal sulphides have received considerable attention.309–314Altering the size of the particle affects the electronic structure of the solid, particularly band gaps, which are tunable by changing the particle size. This offers chemists and material scientists the opportunity to change the electronic and chemical properties of materials simply by controlling their particle shape. This discovery opened the field for many new application areas.315–337
Mechanochemical synthesis has been applied for preparation of a wide range of sulphidic nanocrystals.6,338–345
The acetate route has been used for synthesis of MeS (Me = Zn, Cd and Pb) nanocrystals.343,346–348 MeS nanoparticles were prepared by high-energy milling according to the reaction
| (CH3COO)2Me·xH2O + Na2S·9H2O → MeS + 2CH3COONa + xH2O | (7) |
ZnS nanoparticles were prepared by high–energy milling of zinc acetate and sodium sulphide according to the above-described reaction. The reaction is thermodynamically feasible at ambient temperature, as the enthalpy change is negative. The value ΔH°298 −171 kJ mol−1 was calculated from thermodynamic data.349 The properties of mechanochemically synthesized ZnS(M) were compared with the chemically precipitated ZnS(C).350 The XRD analysis of the ZnS(M) pattern shows mainly the reflections of the cubic phase, which is also supported by relative intensity. The higher background on the XRD pattern of ZnS(M) implies the formation of some amorphous material. The amorphization is in fact a highly distorted periodicity of lattice elements, and it is often characterized as a short-range order in contrast to the long-range order of a fully crystalline structure.
The Warren-Averbach and Williamson-Hall analysis provides detailed information regarding to crystallite size, lattice strain and their distributions.351 Regarding the ZnS(M), the presence of non-zero slope and intercept reveals that both size and strain components exist in the sample. The volume weighted crystallite size and maximum lattice strain calculated are 4 nm and 7.5 × 10−2, respectively. The specific surface area of ZnS(C) (7 m2 g−1) is lower in comparison with ZnS(M), where this value attains 97–128 m2 g−1 with an increase with milling time, which is a further advantage of the mechanochemically synthesized ZnS nanoparticles. Surface morphology of the ZnS(M) with estimated size from 10–20 nm is depicted in Fig. 18a. From the figure it is clear that ZnS(M) forms irregular particles.
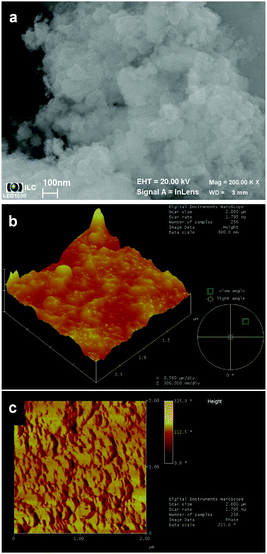 | ||
| Fig. 18 SEM (a) and AFM analysis (b and c) of mechanochemically synthesized ZnS(M).350 | ||
From the surface analysis using SEM it is revealed that there is a homogeneous distribution of the particles and the surface is smooth. Individual nanoparticles have a tendency to form nanoparticle agglomerates during the milling process and both entities can be clearly seen. In order to analyze the surface characteristics in more detail, sampling with AFM in contact mode was used. Fig. 18b shows a 3D representation of the image obtained from the surface of the ZnS(M) nanoparticles. A homogeneous distribution of crystalline domains can be observed from the image. The deflection image of the ZnS(M) nanoparticles surface is shown in Fig. 18c, from which the crystalline distribution was observed to be uniform and regular. From the comparison of the deflection image (Fig. 18c) and the altitude topography (Fig. 18b) it is observed that the crystallites were of the same size. HRTEM technique allows determination of the size of the nanoparticles,352 the type of structures produced,353 and also the possible morphologies.354 In Fig. 19 four different micrographs of ZnS are shown. In Fig. 19a, an area of 16 × 16 nm is observed, where several clusters are clearly identified, and the sizes of three of them were determined to be 2.6, 3.7 and 3.4 nm. In fact, the corresponding fast Fourier transformation (FFT) technique denotes a polycrystalline material, which must be composed of the nanocrystals. Higher magnification allows determination of the lattice distances in the material (Fig. 19b). It is possible to find a square contrast in the center of the micrograph with interplanar distances of 0.27 and 0.28 nm, which implies a region with an axis zone near to the (001). Fig. 19c shows a well-defined cluster of 4 nm with the hexagonal profile and rhombic internal contrast that are characteristic for a truncated octahedron particle observed in the (011) zone axis. The HRTEM images allow defects to be found in the nanocrystalline material, as marked with an arrow in Fig. 19d, which have been reported for the mechanochemically synthesized nanostructures. These defects can be the cause of the processes that induce the reduction of the size of nanocrystals and the corresponding properties.
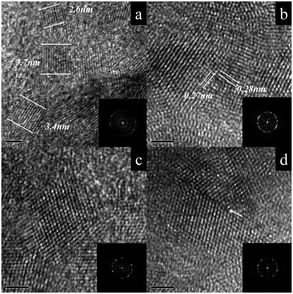 | ||
| Fig. 19 HRTEM micrographs of mechanochemically synthesized ZnS(M): (a) identification of nanoparticle with size around 3 nm, (b) determination of structure with help of the interplanar distance measurement, (c) hexagonal profiles for fcc-like nanoparticles, (d) example of fracture induced in the nanoparticles.350 | ||
Ref. 355 describes structural and surface properties of the CdS nanoparticles synthesized in the planetary mill. The peaks associated with the (111), (220) and (311) planes of the cubic structure were clearly identified. Cadmium sulphide nanocrystallites with particle sizes of 2 nm synthesized in the planetary laboratory mill show a quantum-size effect. The mechanochemically synthesized CdS nanoparticles have different physicochemical properties than chemically synthesized CdS.
ZnxCd1−xS nanoparticles with different compositions were synthesized by high-energy milling in a planetary laboratory mill.356,357 Mechanochemical synthesis produced uniform particles with respect to the microstructure characteristics as a function of milling time. Crystalline sizes of approximately 2 nm were calculated from XRD analysis. The calculated lattice parameters of mixed crystals were found to linearly depend on the composition of the ZnxCd1−xS nanoparticles. The SEM images show aggregates of small cauliflower-like nanoparticles of various sizes, but lower than 50 nm.
PbS nanocrystals have been successfully prepared using surfactant-assisted mechanochemical synthesis.358,359 The XRD patterns confirmed the presence of galena PbS (JCPDS 5-592) whatever treatment conditions were applied. The strong observable peaks in XRD patterns indicate the highly crystalline nature in formation of PbS nanostructures. Surface-weighted crystallite sizes of 3, 8, 11 and 19 nm have been calculated from XRD data using the Warren-Averbach method and confirmed by high-resolution SEM images for surfactant-free and three different surfactant-assisted mechanochemical syntheses. The surfactants facilitate the preferential growth of PbS nanocrystals in the (200) direction.
The mechanochemical synthesis (MCS) of Sb2S3 and Bi2S3 nanoparticles has been studied, starting from the corresponding metals and sulphur and using high-energy milling.360 Both sulphides are semiconductors with interesting thermoelectric properties. The direct band gap of antimony sulphide is 1.78–2.50 eV, and it has interesting high photosensitivity and high thermoelectric power.
Antimony sulphide was prepared according to the reaction
| 2Sb + 3S → Sb2S3 | (8) |
The reaction is thermodynamically feasible, as the enthalpy change is negative, ΔH°298 = −169.4 kJ mol−1.361 XRD patterns illustrating the mechanochemical synthesis of Sb2S3 nanopowder are shown in Fig. 20. Pattern (a) shows the starting mixture of antimony (JCPDS 35-732, rhombohedral) and sulphur (JCPDS 8-247, orthorhombic). Pattern (b) shows an intermediate state that is a mixture of the initial and final phases. After 60 min of milling (pattern (c)) only Sb2S3 (JCPDS 42-1393, orthorhombic) is detected, showing that the reaction is complete.
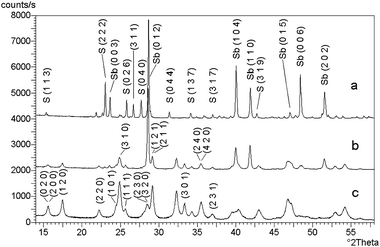 | ||
| Fig. 20 XRD patterns of Sb-S powders before milling (a) and after 15 min (b) and 60 min (c) of milling. The lines labelled by their Miller indexes alone belong to Sb2S3. Reprinted with permission from ref. 360. Copyright 2013, Elsevier. | ||
Bismuth and sulphur react according to the equation
| 2Bi + 3S → Bi2S3 | (9) |
The enthalpy change for this reaction is similarly negative, ΔH°298 = −176.6 kJ mol−1.361 The progress of the mechanochemical synthesis of Bi2S3 nanoparticles is illustrated by the XRD patterns shown in Fig. 21. The process is similarly straightforward, with Bi2S3 (JCPDS 17-320, orthorhombic) being the only solid product (pattern d). In the starting material (pattern a) the peaks of Bi (JCPDS 44-1246, rhombohedral) and S (JCPDS 8-247, orthorhombic) are seen, while patterns (b) and (c) represent intermediate states with different ratios of the initial and product phases.
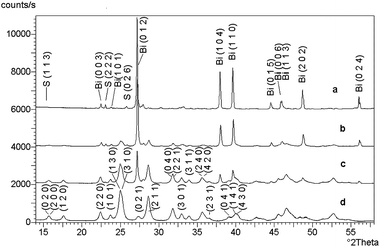 | ||
| Fig. 21 XRD patterns of Bi-S powders before milling (a) and after 4 min (b), 7.5 min (c), and 60 min (d) of milling. The lines labelled by their Miller indexes alone belong to Bi2S3. Reprinted with permission from ref. 360. Copyright 2013, Elsevier. | ||
In ref. 360, the structural, surface and morphology changes as well as kinetics of the mechanochemical synthesis of Sb2S3 and Bi2S3 are presented. The orthorhombic Sb2S3 and orthorhombic Bi2S3 nanoparticles are the only product of the mechanochemical synthesis. XRD patterns reveal the highly crystalline nature of Sb2S3 and Bi2S3 nanoparticles. Normalized XRD data were utilized for a solid-state kinetics calculations. Bi2S3 is formed faster than Sb2S3. The values of specific surface area of mechanochemically synthesized Sb2S3 and Bi2S3 nanoparticles were approximately 13 times and 30 times higher than the corresponding value of the initial mixture Sb + S and Bi + S, respectively. The particle size is about 30 nm for Sb2S3 and 24 nm for Bi2S3. The mechanochemical synthesis of Sb2S3 and Bi2S3 nanoparticles from the corresponding metals with sulphur powder by high-energy milling is suitable for a large-scale mechanochemical preparation.
4.5 Selenides
In general, the conventional (ceramic) methods of metal selenide preparation require high temperatures. However, the reactions leading to selenides can be carried out at ambient temperature in a planetary ball mill. AIIBVI (ZnSe), AIVBVI (PbSe, SnSe), AIVB2VI (SnSe2), AVBVI (BiSe), and A2VB3VI (Bi2Se3) types of binary selenide semiconductors were mechanochemically synthesized by high-energy milling of corresponding powder precursors according to the thermodynamically feasible reactions shown in Table 3.Table 3 Mechanochemical synthesis of metal selenides
| Reaction | ΔH°298/kJ mol−1 |
|---|---|
| Zn + Se →ZnSe | −159 |
| Pb + Se → PbSe | −100 |
| Sn + Se → SnSe | −89 |
| Sn + 2Se → SnSe2 | −121 |
| Bi + Se → BiSe | −53 |
| 2Bi + 3Se → Bi2Se3 | −140 |
AB type – ZnSe, PbSe, SnSe, BiSe. Zinc selenide (ZnSe) belongs to the group of II–VI semiconductors. These materials are of interest because of the possibility of their utilization in various optoelectronic applications such as blue light-emitting diodes, laser diodes, solar cells, optical recording materials and thermoelectric cooling materials.362 Lead selenide (PbSe) and tin selenide (SnSe) are semiconductors of the IV–VI group, having a wide range of applications including optical switching, optical computing, telecommunication components, photovoltaics, thermoelectrics, photodetectors, infrared detectors, mid-infrared lasers, solar or photoelectrical cells, chemical sensors, and holographic recording systems.363,364 Bismuth selenide (BiSe) belongs to the V–VI binary semiconductor group and is interesting because of its optical, electrical and thermoelectric properties.365,366
The X-ray diffraction patterns of the mechanochemically synthesized AB-type semiconductors with corresponding time of mechanochemical synthesis are shown in Fig. 22. The peaks were identified based on the JCPDS card and correspond to zinc selenide (stilleite, 15–0105), lead selenide (clausthalite, 06–0354), tin selenide (32–1382) and bismuth selenide (nevskite, 29–246). Synthesized samples were ultrasonically de-agglomerated for TEM investigations (Fig. 23). Most of the nanocrystals were easily separated from the agglomerates while some of them remained captured in the larger clusters with the size from about 100 to 500 nm. ZnSe nanoparticles (Fig. 23a) were of irregular shape with a size of about 5–20 nm. In close proximity to the clusters the needle structures are visible; EDX analysis detected that the needles consist of selenium.367 PbSe nanocrystals were idiomorphic and their size ranged from a few nanometers up to 80 nm (Fig. 23b). The prevailing crystallographic form of the crystallites is a cube {100} which is occasionally modified by narrow rhombic-dodecahedral faces {101} truncating the cube edges.368 TEM analysis of the mechanochemically synthesized SnSe sample confirmed the presence of thick clusters of un-reacted Sn- and Se-rich amorphous phase including numerous large idiomorphic SnSe crystals with sizes up to 80 nm (Fig. 23c). The measurement of the lattice spacings from the high-resolution TEM image confirmed the presence of orthorhombic SnSe. The smallest crystals in this sample have sizes in the range between 2–8 nm.369
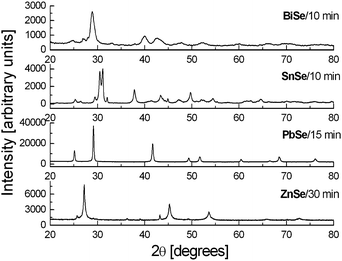 | ||
| Fig. 22 X-ray diffraction patterns of mechanochemically synthesized AB-type selenide semiconductors. | ||
 | ||
| Fig. 23 TEM images of mechanochemically synthesized ZnSe (A), PbSe (B), and SnSe (C) semiconductor nanoparticles. | ||
The absorption behavior of mechanochemically synthesized ZnSe, PbSe and SnSe was studied by UV-Vis-NIR spectroscopy (Fig. 24). Optical absorption spectra of ZnSe nanoparticles synthesized for 30 min and PbSe nanoparticles synthesized for 15 min showed the expected size-dependent effect of quantum confinement and exerted a blue shift from the direct band-gap energies 2.67 eV and 0.28 eV characteristic for bulk ZnSe and PbSe crystals, respectively.370,371 The spectrum of ZnSe illustrates an extended absorption edge at about 548 nm that corresponds to 2.27 eV.372 The spectrum of PbSe illustrates a small extended absorption edge at about 1032 nm that corresponds to 1.2 eV.368
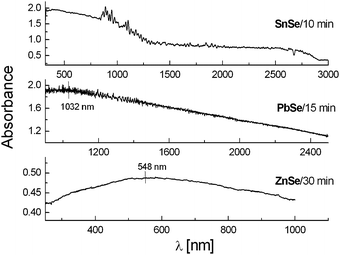 | ||
| Fig. 24 UV-Vis-NIR optical absorption spectra of mechanochemically synthesized AB-type semiconductors. | ||
This effect can be attributed to the existence of very small ZnSe and PbSe nanoparticles, which are agglomerated into the big clusters. The absorption spectrum of SnSe synthesized for 10 min demonstrated good absorption in the visible region without the evidence of a quantum confinement effect.
AB2 type – SnSe2. Tin diselenide (SnSe2) belongs to the group of II–VI semiconductors with layered structure which have brilliant application prospects due to their optical and electrical properties and can be applied in film electrodes, infrared optoelectronic devices, thermoelectric refrigerators and solar cells. The XRD pattern of SnSe2 synthesized for 100 min is given in Fig. 25. The diffraction peaks belonging to the reaction product are consistent with the hexagonal SnSe2 phase (JCPDS PDF 89-3197).373 The maximum value of the specific surface area, 1.39 m2 g−1, suggests the completion of the mechanochemical event after 100 min of milling. TEM study of SnSe2 confirmed that the sample is composed of the hexagonal modification. Fig. 26a and c show typical SnSe2 crystals forming thick barrel-shaped platelets with particle sizes ranging from a few nm to more than 100 nm. The morphology of the SnSe2 nanocrystals is bipyramidal-hexagonal (space group P![[3 with combining macron]](./Hallmarks_files/char_0033_0304.gif) m1). Diffraction rings in the SAED pattern (Fig. 26b of SnSe2) match the d-values for hexagonal SnSe2 phase (JCPDS PDF 89-3197). From UV-Vis-NIR measurements, the indirect and direct bandgap values 1.0 eV and 1.25 eV for mechanochemically synthesized SnSe2 nanocrystals were derived. However, the samples showed different types of transitions in the lattice. The results of 119Sn MAS NMR and Mössbauer spectroscopy confirmed that a one-step mechanochemical synthesis yields almost pure SnSe2.373
m1). Diffraction rings in the SAED pattern (Fig. 26b of SnSe2) match the d-values for hexagonal SnSe2 phase (JCPDS PDF 89-3197). From UV-Vis-NIR measurements, the indirect and direct bandgap values 1.0 eV and 1.25 eV for mechanochemically synthesized SnSe2 nanocrystals were derived. However, the samples showed different types of transitions in the lattice. The results of 119Sn MAS NMR and Mössbauer spectroscopy confirmed that a one-step mechanochemical synthesis yields almost pure SnSe2.373
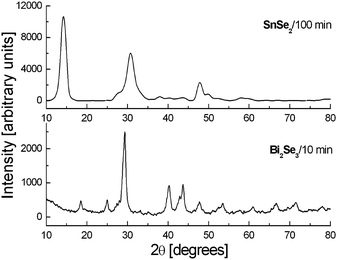 | ||
| Fig. 25 X-ray diffraction patterns of AB2 and A2B3 types of mechanochemically synthesized selenide semiconductors. | ||
 | ||
| Fig. 26 TEM images of mechanochemically synthesized SnSe2 and Bi2Se3 nanoparticles. | ||
A2B3 type – Bi2Se3. Bismuth triselenide (Bi2Se3) is a member of the V–VI binary semiconductor group which has attracted considerable attention due to their characteristic anisotropic layered structure and semiconducting properties.374 This material has therefore found many applications in the field of optical recording systems, as laser materials, optical filters, sensors, solar cells, strain gauges, narrow bandgap semiconductors, electromechanical and thermo-electric devices, and topological insulators. The XRD peaks of Bi2Se3 synthesized for 10 min (Fig. 25) were identified on the basis of the JCPDS card 33–214, and it was found that they correspond to paraguanajuatite – Bi2Se3 phase.375 The maximum value of the specific surface area, 1.47 m2 g−1, was reached for Bi2Se3 after 10 min of milling and evidenced completion of mechanochemical synthesis. TEM analysis revealed two types of grains: larger grains with diameters up to 300 nm and nanosized particles with sizes down to about 5 nm. Bi2Se3 nanocrystallites are shown in Fig. 26a. The SAED pattern recorded from these grains showed typical diffraction rings (Fig. 26b) of Bi2Se3 that can be indexed with the hexagonal Bi2Se3 phase (JCPDS PDF card 33–214). A HRTEM image of a single nanocrystallite (Fig. 26c) of Bi2Se3 revealed the characteristic layered structure of the Bi2Se3.375
According to the literature, two different values for the minimum energy gap are reported for bulk Bi2Se3 – 0.35 and 0.16 eV.376 The optical band gap energy of mechanochemically synthesized Bi2Se3, 1.37 eV, was obtained on the basis of the recorded and derived absorption spectrum in the UV–Vis spectral region. The higher band gap value of Bi2Se3 in comparison with the bulk one indicates a blue shift phenomenon due to the quantum size effect. This effect can be attributed to the existence of very small bismuth selenide nanoparticles agglomerated into the big clusters.375
4.6 Carbides, nitrides, borides and silicides
Non-oxide refractory carbides, nitrides, borides and silicides are candidates for high-temperature structural applications. The conventional synthesis of these compounds involves high-temperature treatments that extend over long periods of time, which hinders control over the purity, homogeneity, particle size and particle size distribution in the final compounds. Increasing engineering demands, such as more reliable high-temperature stability and better mechanical properties, require the use of nanosized powders. This has forced the development of new synthetic routes. Nevertheless, temperature often remains a primary reaction-controlling and limiting parameter. In this context, mechanochemistry appears to be an attractive and promising option because it is a relatively simple method that enables cost-effective mass production at room temperature.Mechanochemical synthesis from elemental mixtures. Among the mechanochemical reactions performed to produce these refractory compounds, the direct formation reaction from the elements has been frequently used. The feasibility of this procedure was proposed by Matteazzi and Le Caër377 for metal carbides and has been employed by many other research groups. For example, carbides such as c-TiC,378–388 c-ZrC,388–390 c-HfC,391 c-VC,392 o-V2C,392 c-NbC,378,388,393 c-TaC,394 h-WC,388,395,396 c-Cr23C6,397 h-Cr7C3,397 o-Cr3C2397 and c-SiC398–402 have been obtained by milling powder mixtures that contain the corresponding element and carbon in the required stoichiometric ratio in an inert atmosphere. The mechanochemical synthesis of c-TiC has also been successfully performed in vacuum and H2 atmospheres.384 In this latter atmosphere, the presence of the intermediate c-TiH2, which has a similar structure to that of c-TiC, was observed during milling. Special attention must be paid to avoid this phase in the product.
Graphite has been widely employed as the carbon source for these compounds, but carbon black,395 activated carbon,381 and even carbon fibres and nanotubes383 have also been used. Various carbon-rich organic media, such as deoiled asphalt,382 petroleum coke,380 toluene,388 hexane393 and heptane,387 which decompose during milling to release atomic carbon, have also been proposed as raw materials. When the organic substance contains hydrogen, precautions similar to those employed when using a H2 atmosphere should be used to avoid c-TiH2.387
Similarly, the synthesis of nitrides such as c-TiN,403–407 c-ZrN,404,405,408 c-HfN,409 c-VN,410,411 h-NbN (δ′),412 c-NbN (δ),405,413 h-Ta2N,412 c-TaN (δ),405 c-Mo2N (γ)414 and c-CrN405,415 has been performed by milling the transition metal powders under N2 atmosphere. As long as excess nitrogen is available to complete the chemical transformation, the reaction can occur at a low gas pressure, although increasing the pressure accelerates the process.407 It has been proposed that c-TiN can be obtained by milling Ti in air,416 but some authors have demonstrated that the obtained cubic phase actually corresponds to a nitrogen-rich titanium oxynitride.417,418
The cubic γ-Mo2N, δ-NbN and δ-TaN obtained at room temperature are high-temperature forms and are therefore metastable phases. Furthermore, the hexagonal δ′-NbN is also a metastable phase, which is reported to occur in the transition region between δ-NbN (c) and ε-NbN (h), the high- and room-temperature forms, respectively. Ogino et al.405 reported the formation of a metastable c-MoN phase by milling Mo in N2, although the XRD pattern presented in their article is similar to that shown by Lucks et al.414 that corresponded to the stable c-Mo2N phase, which was obtained in a similar way.
By milling Ta in N2, Liu et al.412 obtained the stable h-Ta2N, but Ogino et al.405 observed the formation of the metastable δ-TaN. In the Nb–N system, Liu et al.412 obtained the metastable δ′-NbN, whereas Ogino et al.405 and El-Eskandarany et al.413 observed the metastable δ-NbN after the full nitriding of Nb. Note that the metastable δ′-NbN has the same structure as the stable h-Ta2N. The different obtained products were apparently related to the intensity of the milling process. Consequently, Ogino et al. obtained a mixture of δ-NbN and δ′-NbN rather than a single phase δ-NbN by reducing the frequency of the vibrational mill they used.405
Other studies have demonstrated that the mechanochemical syntheses of c-TiN,419,420 c-ZrN,421 c-VN422 and h-AlN423 can be achieved by milling the powdered metals in a dry ammonia atmosphere and that the milling process is faster than in nitrogen.420 During the synthesis of c-TiN and c-ZrN, the formation of cubic hydrides was observed during the early stages of milling. These phases were transformed into the nitride phases after further milling.419,421 It has been observed that the c-TiN powders exerted an important extent of non-stoichiometry, with some of the vacancies most likely occupied by hydrogen atoms.420
Some authors have proposed the use of solid nitrogenous organic compounds to improve the contact of the reactants and to avoid the technical problems associated with the supply of the reactive gas in the solid–gas mechanochemical reactions. For example, c-TiN has been synthesised by milling Ti and urea,424 and h-AlN has been produced by milling Al and melamine.425
Many of the refractory carbides and nitrides are non-stoichiometric compounds and are stable within a wide compositional range. It has been observed that during the early stages of milling, these materials can be obtained with a high number of vacancies; however, by prolonging the milling process, the stoichiometric composition is generally obtained.
Refractory borides, including h-TiB2,426,427 h-ZrB2,428 h-HfB2,428 h-VB2,429 o-NbB,430 h-NbB2430,431 and h-MoB2432 have been obtained by milling stoichiometric elemental mixtures in an inert atmosphere. The synthesis of h-TiB2 by milling Ti in air was also reported, but details of the oxygen contamination were not provided in this study.433 According to the Mo-B phase diagram, the h-MoB2 phase is stable above 1517 °C. Mechanochemistry allows this metastable phase to be obtained at room temperature. It can also be produced by heating the r-Mo2B5 phase to high temperatures followed by quenching.
Different refractory silicides, such as h-Ti5Si3,434–436 t-Mo5Si3,437 o-TiSi2 (C54),436 o-TiSi2 (C49),434 o-ZrSi2 (C49),389a,b h-VSi2 (C40),438,439 h-NbSi2 (C40),439,440 h-TaSi2 (C40),394,439 h-CrSi2 (C40),439 t-MoSi2 (C11b),439,441,442 h-MoSi2 (C40)443,444 and t-WSi2 (C11b)439,445 have also been obtained as single phases by milling stoichiometric elemental mixtures in an inert atmosphere. C40-MoSi2, which is a high temperature phase, and C49-TiSi2, which does not appear in the Ti–Si phase diagram, are both metastable phases. Some considerations about these phases will be indicated later.
Mechanochemical synthesis is interesting for the formation of solid solutions and complex compounds because it can avoid solubility variations between the different components to provide homogeneous products more readily than the thermal methods. Furthermore, the stoichiometry of the final product is well correlated with the composition of the starting mixture. Among others, h-W1−xAlxC (0.33 < x < 0.86),446 c-TixV1−xNy,447 c-Ti1−xAlxN (0 < x < 0.5),448 c-Nb0.5Al0.5N,449 c-TiCxN1−x,450 c-NbCxN1−x,451 c-TaCxN1−x,451 c-HfCxN1−x,451 c-TiyZr1−yCxN1−x,452 c-TiyHf1−yCxN1−x,452 c-TiyV1−yCxN1−x,452 c-TiyNb1−yCxN1−x,453 c-TiyTa1−yCxN1−x,452 c-ZrB1−xNx,454 h-Hf1−xZrxB2,455 h-Ti1−xZrxB2428 and h-Ti1−xHfxB2 (0.5 < x < 1)428 solid solutions were obtained by milling the stoichiometric elemental mixtures in the appropriate atmosphere.
These refractory compounds, especially those from the IVB and VB groups, have large negative enthalpies of formation. The majority of these compounds have been produced by self-propagating high-temperature synthesis (SHS). The mechanochemical synthesis from elemental mixtures is also characterised by a considerable heat release. A combustion-like process similar to SHS, which is called a mechanically induced self-sustaining reaction (MSR) has been frequently reported during milling.378,381,384–386,389,391,406,407,426–428,435,436,439,441,442,447,450–453,455 In these cases, the majority of the reactants instantly transform into the products after a critical milling time (the ignition time). Self-sustaining reactions have never been observed for the less exothermic systems (e.g., Cr-C, W-C, Si-C and W-Si). In this case, the conversion into products gradually proceeds with the milling time as a result of diffusional solid state reactions between the starting phases.
Naturally, the occurrence of MSR effect during milling has not been reported because the products were generally inspected after long milling times, after ignition was likely to have occurred.393,434 The limited propagation of the self-sustained reaction in some powder mixtures can render the seemingly gradual compound formation. Although the formation of nitrides is an extremely exothermic reaction, MSR effects during the solid–gas mechanochemical synthesis of these compounds have been barely observed,406,407,447 and an important amount of product formed by diffusion was always observed before ignition. In general, when solid–gas reactions are performed, significantly longer milling times are needed than when only solid phases are involved. Calka et al.456 have developed a method that combines mechanical milling with an electrical discharge to enhance the reaction rate and reduce total milling time. Full conversion into c-TiN was observed after milling Ti in N2 for only 30 min.
Both reaction mechanisms, MSR or gradual, have been frequently observed for the same reactant mixture, and the transition between both regimes was related to the milling intensity. Deidda et al.457 have shown that the synthesis of c-TaC from mixtures of Ta/C can proceed via MSR process and that the ignition time decreased with increasing impact energy. Furthermore, the authors observed the gradual formation of c-TaC at low impact energy. Roldan et al.447 reported that different reaction mechanisms occurred during the synthesis of the c-TixV1−xNy solid solution by milling Ti and V in N2 depending on the composition of the starting mixture. The authors observed the MSR process for the Ti-rich starting mixtures and diffusional mechanism for the V-rich starting mixtures as a consequence of higher exothermicity with increasing Ti content in the starting mixture.
Different final products were occasionally obtained depending on the mechanochemical reaction mechanism. In the synthesis of MoSi2, the stable C11b-MoSi2 was obtained when MSR process was induced during milling of the elemental mixture of reactants.439,441 However, the metastable C40-MoSi2 only formed through a diffusional mechanism with lower milling intensity.444 It was also observed that the C11b-MoSi2 obtained by MSR can be transformed into the C40-MoSi2 phase by further milling.442 Similarly, the stable C54-TiSi2 was obtained when the MSR process was achieved during synthesis.436 When a diffusional mechanism was followed, a mixture of metastable C49-TiSi2 and stable C54-TiSi2 was generally observed. Yan et al.434 could obtain single-phase C49-TiSi2 using a milling procedure that was interrupted every 15 min to cool the vials for 15 min. These examples demonstrate that the MSR process drives the formation of stable phases, and, therefore, if the goal is the synthesis of a metastable phase, it is compulsory to avoid MSR by selecting appropriate milling parameters.
Mechanochemical synthesis by displacement reactions. Mechanochemical processes based on displacement reactions, which involve a metal powder and the oxide of a less reducing metal, have also been proposed for the mechanochemical synthesis of refractory compounds. This synthetic route is of particular interest because readily available and less expensive materials can be used. For example, r-B4C was obtained by milling B2O3 and Mg in the presence of carbon.458 In fact, the synthesis of r-B4C involves two coupled reactions, where the B obtained by the reduction of B2O3 with Mg is the reactant for the second reaction with C to form r-B4C. MgO, the by-product of the first reaction, is present in the final product but can be removed by acid leaching. The leaching treatment also has the advantage of eliminating other contaminants, such as iron from the milling media, but it should be carefully controlled because the partial dissolution of the product, which becomes more pronounced with decreasing particle size, can significantly reduce the final yield. c-VC,459 h-WC460,461 and c-SiC460 were also produced from V2O5–Mg–C, WO3–Mg–C and SiO2–Mg–C mixtures, respectively. For the synthesis of h-WC and c-SiC, the use of a hydrogen or hydrocarbon atmosphere has been proposed to increase the yield and to avoid the formation of secondary phases in the product.460
Similarly, h-TiB2462–464 and h-ZrB2465,466 have been obtained by milling TiO2–B2O3–Mg and ZrO2–B2O3–Mg mixtures, respectively. In these cases, the process involves three different reactions, which include the reduction of both oxides to produce elemental Ti or Zr and B that subsequently react to yield the final product. In general, Mg is used in excess over the stoichiometric amount to compensate for surface oxidation of Mg and residual oxygen adsorbed onto reactant surfaces, although in some cases residual oxides always remained in the final product.465,466
The magnesiothermic reductions are strongly exothermic, and the process generally proceeds in a self-sustaining manner. The individual coupled reactions are not distinguishable, and only one MSR effect corresponding to the global reaction was observed. However, this synthetic route can also follow a diffusional mechanism. For example, El-Eskandarany461 did not detect a self-sustaining reaction during the synthesis of h-WC, and for intermediate milling times a mixture of reactants and products of the coupled reactions was observed.
Other mechanochemical routes based on exothermic double-displacement reactions have been proposed with the aim of reducing the milling time to attain full conversion. For example, c-ZrN was obtained after only 7 min by milling a mixture of Li3N and ZrCl4.467 h-TiB2 and h-VB2468 were synthesised from mixtures of TiCl3, LiBH4 and LiH and VCl3, LiBH4 and LiH, respectively, and c-NbC469 from NbCl5 and CaC2. In these processes, the obtained by-products (LiCl or CaCl2) were removed by rinsing in mild conditions. These by-products help to reduce agglomeration, and in the synthesis of h-TiB2 and h-VB2, Kim et al.468 added a critical amount of LiCl to the reactant mixture to suppress the MSR effect and to act as a process control agent (PCA) and in the end they obtained nanoparticles with a low agglomeration level.
Milling time. The MSR processes allow a high yield of product to be obtained in a short time, but the reaction is often incomplete after propagation. The extent of the conversion depends on the reactant mixture and the milling conditions. Prolonging the milling time for a certain period is required to attain full conversion. Furthermore, the microstructure of the product obtained immediately after the self-sustaining reaction is characterised by submicrometric agglomerated particles with a large size distribution. These agglomerates occasionally exhibit a sintered appearance as a consequence of the heat released during the highly exothermic reaction. Therefore, if the goal is to obtain nanosized particles with a narrow size distribution, it is absolutely mandatory to prolong milling for a significant amount of time.
When the mechanochemical reaction is controlled by diffusion, the conversion of reactants into the product is gradual, and the time required to achieve full conversion is significantly longer compared to the MSR processes. However, due to the special synthetic conditions, where the reaction progresses by small diffusion paths at the interfaces of the reactants during impacts that are phenomena of short duration, a new phase with a nanometric character is formed,411 and the milling process can be terminated once conversion is reached. No further milling is required.
The milling time required to obtain a given refractory compound is a function of the reactants, the reaction mechanism, the specific equipment and the operating conditions used to perform the mechanochemical synthesis. For example, during the synthesis of c-HfN by milling Hf in N2,409 it was demonstrated that the degree of nitridation depended on the total supplied energy dose, irrespective of the single impact energy. With increasing impact energy, a net increase in the rate constant was observed, and the milling time required to attain the same conversion degree was consequently reduced.
A wide range of milling times is observed in the literature for the same compound. For example, if the ignition time is used for comparison, during the synthesis of c-TiC from Ti–C mixtures times ranging from 35 min385 to 70 h381 were observed. Unfortunately, the full set of experimental parameters is often not provided in the articles, and comparing the results is difficult.
Contamination. The mechanochemical synthesis of refractory compounds at the nanoscale, irrespective of the reaction mechanism (MSR or diffusion), requires high-energy conditions and long milling times, typically several hours. Contamination resulting from the wear of the milling media, such as iron from steel balls and vials, can be a considerable drawback that should not be neglected, because if no special care is taken then it can reach high values. Iron impurities can also modify the stoichiometry of the reaction.470 Contamination is often not quantified and is frequently excluded based on XRD measurements, which are not accurate because of the low intensity and considerable broadening of reflections and the high level of background scattering that characterises the XRD patterns of products obtained by milling. For example, Rahaei et al.386 did not observe the presence of iron by XRD during the synthesis of c-TiC using steel milling media, but the chemical quantification revealed iron contamination of 0.09, 0.28, 0.72 and 1.76 wt% after 1, 4, 8 and 16 h of milling, respectively.
Although wet milling eases the formation of nanosized particles with narrow size distributions, the contamination due to abrasion of the milling media is significantly higher compared with dry milling, where the balls and walls of the vials are coated with a protective layer that is expected to prevent contamination. During the synthesis of c-NbC from a mixture of Nb and graphite, Murphy and Courtney393 observed iron contamination of 4.4 wt% after 24 h of milling, whereas this contamination was increased to 18.3 wt% under similar milling conditions when the c-NbC was obtained by milling Nb in hexane.
Oxygen impurities in non-oxide refractory compounds are detrimental for the resulting mechanical properties because these impurities promote coarsening during sintering at temperatures lower than the ones when densification occurs. Oxygen in mechanochemically synthesised powders frequently comes from the initial reactant powders, inadequate control of the milling atmosphere and inappropriate handling of the products after milling. If all of these factors are controlled, the oxygen contamination can be minimised.
In general, contaminants can be reduced to levels that are acceptable for most potential applications (<1 wt%) of the synthesized compounds with appropriate selection of the milling media (using the same material as the powder being milled396 or at least not harmful to its properties), adequately handling the materials and trying to find a suitable compromise between milling intensity and milling time. For example, in c-ZrC powders synthesised from a Zr–C mixture after 72 h of milling, Mahday et al.390 observed low iron and oxygen contents of 0.84 wt% and 0.21 wt%, respectively.
When nanocrystalline mechanochemical products, as confirmed by XRD measurements, are inspected by SEM, submicrometric agglomerate particles are frequently observed. However, if these particles are inspected by HRTEM, it can be observed that they are actually composed of many randomly oriented nanocrystallites, which is confirmed by electron diffraction patterns characterised by sharp, nearly continuous rings. The size of these nanodomains is consistent with the average grain size estimated from the XRD patterns. Prolonging the milling time can help the powders become more uniform in shape and size,390 but it can also induce the partial amorphisation of the product.412,414,430 The use of PCA can allow smaller agglomerates with narrower size distribution to be obtained, as shown during the mechanochemical synthesis of c-SiC by the addition of 2 wt% of NH4Cl to the initial Si–C reactant mixture.402 But, as stated above, the use of long milling times or PCA can be a source of contamination.
5. Multi-phase nanosystems
5.1 Metal-ceramic nanocomposites
Literature reviews14,186,471 highlight some dependences of formation, structure, and properties of mechanochemically synthesized composites in mostly metallic systems. This section considers two groups of two-component systems: light metals with flaky second component Me/X (Me = Al, Mg, Ti, Fe, Si; X = C, h-BN) and energy-saturated systems with high specific heat of interaction Me1/Y (Me1 = Al, Mg, Si; Y = MoO3, (C2F2)n, etc.).Initial stage of mechanochemical synthesis. Formation of nanocomposites is usually a necessary intermediate stage of mechanochemical synthesis. For example in the process of mechanochemical synthesis of Al–C system,173,472a,b one can distinguish three intermediate stages (Fig. 27). During the first stage, there is an independent disintegration of components (Al and C) and their mixing. The growth of specific surface area during this stage is due to destruction of graphite. At the same time, there is a reduction in the size of coherent scattering regions (CSR) for Al. During the second stage, there is surface interaction of carbon with aluminum. As a result, Al becomes more brittle and falls apart to nano-sized structures. This leads to the formation of composite structures that are made of Al nanoparticles separated by layers of carbon.472c Then, the molecular-dense compaction of the mixture takes place, which is accompanied by a significant decrease of the specific surface area. During the third stage, practically all atoms participate in chemical interaction of metal with carbon. This process is accompanied by the crystallization of the synthesized product.
![Three stages of mechanochemical synthesis of the example reaction Al + C → Al3C4. Stage 1: independent destruction of components, stage 2: nanocomposite formation, stage 3: synthesis and crystallization of product. The dependence of concentration of [Al4C3] (1) and [Al–C] bonds (2), specific surface area S of mixture (3) and CSR for Al (4) on the duration t of mechanical treatment. Al-30 wt% C. The maximum S = 96 m2 g−1.](./Hallmarks_files/c3cs35468g-f27.gif) | ||
| Fig. 27 Three stages of mechanochemical synthesis of the example reaction Al + C → Al3C4. Stage 1: independent destruction of components, stage 2: nanocomposite formation, stage 3: synthesis and crystallization of product. The dependence of concentration of [Al4C3] (1) and [Al–C] bonds (2), specific surface area S of mixture (3) and CSR for Al (4) on the duration t of mechanical treatment. Al-30 wt% C. The maximum S = 96 m2 g−1. | ||
The proposed scheme of stages for nanocomposite formation is valid for a broad spectrum of Me–X systems. The similar data were obtained for systems Fe–C,473 Zr–C,474 Ti–C,475 TiH2–C,476 Mg–C,477 Mo–C,478 Si–C,479 Al–BN and Mg–BN,480 Ti–BN480,481 and Fe–BN.482 General mechanisms are identical for all systems.
Reactivity with external reagents. Formation of new, not oxidized and defective surfaces of metal in nanocomposite materials leads to high reactivity of mechanochemically synthesized composites with different reagents. A special technique483 was designed to prevent self-ignition of nanocomposite Al–C during its first contact with air. It was shown that Al in the composite Al–C interacts with oxygen at significantly lower temperature than in case of the usual sample. Experiments were done under heating at a low rate (10 °C min−1)484 and during a shockwave initiation.173 The preparation of nanocomposites by a mechanochemical method is a usual way for improving the properties of materials used in hydrogen storage.186 As an example, mechanical activation of Mg–C–H2,477 Ti–C475 and Ti–BN481 composites lead to the decrease of hydrogenation temperature. There is a significant interest in a possibility of hydrogen yield for mechanochemically prepared Al–C nanocomposites prepared in the presence of water.485 A detailed analysis of the ways by which it is possible to speed up this reaction via mechanochemical treatment is beyond the scope of this work. An interested reader can be referred to several recent publications486 and an exhaustive review.487 Reactions of mechanically activated Al with ethanol and butanol have been studied as well.173 The analysis of reactivity of mechanochemically activated nanocomposites is also given there.
Mechano-thermal approach. A combination of mechanochemical preparation of nanocomposites with their subsequent heating is one of the commonly used synthetic methods. Creation of a high contact surface area of components and the presence of a high concentration of defects on the surface (and in the volume) of the materials significantly speeds up the synthesis and lowers the reaction temperature. Fig. 28 illustrates the role of mechanochemical activation and formation of nanocomposite on the synthesis of Al4C3.488
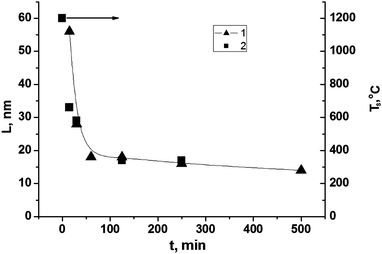 | ||
| Fig. 28 Onset temperature TS for Al4C3 synthesis (2) and size L of CSR for Al (1) as a function of milling time t for Al-15 wt% C mixture. Reprinted with permission from ref. 488. Copyright 2006, Springer. | ||
Thermo-synthesis was completed in a cell of a thermo-analyzer. The progress of the synthesis was controlled by exothermal heat effect and via XRD patterns. Without activation, the synthesis takes place at a temperature 1200 °C, which is significantly higher than the melting temperature of Al. In Fig. 28 the temperature TS at which the exothermal effect starts and CSR of Al are plotted as a function of the duration of mechanical treatment. The formation of Al–C nanocomposites results in a drastic decrease of the Al4C3 synthesis temperature by about 800 °C and a change of the reaction mechanism from liquid to solid phase. It is known that the rate-limiting step for high-temperature synthesis of Al4C3 is the graphite wetting by Al. It was proposed that the TS decrease induced by MA is due to the “forced” mechanochemical wetting of graphite by Al, with partial chemical bonding of components at the surface. Similar data were obtained upon heating mechanically activated Si–C479a and Al–BN480 and their mixtures. In the last case two products (AlN and AlB2) are simultaneously formed.
Mechanochemically activated energetic nanocomposites (MAEC). Recently, there has been an active development of different methods for obtaining energetic materials with the help of nanotechnology. One of the effective and technologically promising methods for obtaining energetic materials is mechanical activation. It was discovered in 2002489 that composites Al/(C2F2)n and Al/MoO3 prepared by mechanochemical activation show a significant increase in the burning rate. This direction of research is currently being explored in Russia490 and in the USA,491 as well as by other groups.492 Up to this point, a possibility of mechanical activation was studied for the following systems: Me1/X (Me1 = Al, Mg, Si, Zr, Ti, Al0.5Mg0.5, MgH2; X = (–C2F2–)n, MoO3, CuO, Fe2O3, Bi2O3, WO3, SrO2, NaNO3, KClO4, NH4ClO4, I). For MAEC Al/MoO3 it was shown that the burning rate increases by three orders of magnitude in comparison with non-activated mixtures. For MAEC Al/(–C2F2–)n and Mg/(–C2F2–)n, a shock wave initiation of the reaction Al + (–C2F2–)n → AlF3 + C led to quasi-detonation regime with speeds of about 1300 m s−1.
During the synthesis of MAEC, it is important to create a maximum contact surface for the components and defects of reagents, but, on the other hand, to prevent chemical interaction between reagents in the process of ball milling. Under optimal conditions of Al(Mg)/(–C2F2–)n mechanical activation, metal particles decrease to 100–150 nm in size and are covered with a layer of polymer (effect of encapsulation). The contact area between components in these composites corresponds to the surface area of metal particles. Submicron metal particles are aggregated into microsized complexes.
In Al(Mg)/MoO3 composites the situation is different. It is relatively easy to make particles with a size of about 60 nm under mechanical activation of MoO3 (Table 2). The presence of the second component (Al, Mg) in the system slows down the process of comminution of MoO3. Therefore, when the Me–MoO3 system was mechanically activated, nanosized MoO3 prepared in preliminary experiments was used. In addition, the presence of MoO3 makes comminution of Mg easier, but not of Al. As a result, after activation these systems are mechanical mixtures of two “independent” components with contact surface area being significantly smaller than the outside surface area of particles. The reduction of the component’s contact surface area is accompanied by worsening of the detonation characteristics.
The effect of the component’s nature on formation and structure of Me/X composites. As can be seen from previous sections, metal-ceramic nanocomposite depends on the nature of the second component. The addition of graphite or h-BN makes dispergation of elastic metals easier and stabilizes 10–30 nm nanostructures in all metals in a form of “raisin in a dough”. In (–C2F2–)n mixtures, particles of Al and Mg decrease in size only to 100–150 nm and get covered with a layer of polymer. Addition of MoO3 prevents the dispergation of Al and leads to formation of a mechanical mixture of components. Such a difference in behavior of two-component mixtures in the processes of mechanical activation can be formally interpreted as different signs of the Rebinder effect. From this perspective, graphite and h-BN are good “mechanochemical” surfactants for the metals, (–C2F2–)n is not so good, and MoO3 is a bad surfactant. Therefore, the considered additives can be written493 sequentially in the order of reduction of their ability to act as a “mechanochemical” surfactants: h-BN > (–CF2–CF2–)n > MoO3 (Mg) > MoO3 (Al). For more detailed analysis of the role of surface forces during mechanical activation of two-component systems further research is required.
However, not all the examples given above can be considered composite materials, but in any case the presence of other phases is very important for their synthesis.
5.2 Further studies
Multicomponent nanosystems can be classified as nanostructured materials which are bulk solids with a nanometer-scale microstructure.194 A significant fraction of atoms of nanostructured materials are in defect environments (grain boundaries, interfaces, interphases, triple junctions). The volume fraction associated with grain boundaries is for instance20% for 10 nm grain with grain boundary thickness of 0.7 nm.494 For preparation of nanostructured materials the term mechanical alloying (MA) has been introduced. MA is a process consisting of a mechanical mixing of elementary components at the nanoscale. A long duration of such a mechanical process is expected to lead to the formation of a stable end product.194 In the MA process of ductile powder mixtures of A and B, particles are trapped between colliding balls or between ball and vial and are subjected to a severe plastic deformation which exceeds their mechanical strength. This is accompanied by a temperature rise. The waiting period between such efficient trapping events is typically of the order of tens to hundreds of seconds.495a,b Particles are repeatedly flattened, fractured and welded. Fracture and welding are the two basic events which ensure permanent exchange of matter between particles and mixing of the various elements of milled powders. As a result, a layer structure with alternating A and B layers is formed, progressively refined and convoluted. Five typical stages of MA of ductile powder mixtures are shown in Fig. 29. | ||
| Fig. 29 Steps of powder evolution during mechanical alloying: 1 – initial powders, 2 – flattering, 3 – welding, 4 – 3D welding and fracturing, 5 – stable end product.194 | ||
A balance between coalescence and fragmentation leading to a rather stable particle size is achieved during MA. The mixture of constituents finally becomes homogeneous and the elements are mixed on an atomic scale.
Originally, MA started in the early sixties at the INCO research laboratories in the USA because of the necessity of developing superalloys based on nickel and aluminum for gas turbine applications and other heat-resistant alloys for the aerospace industry. The alloy was hardened by milling the alloys together with relatively low percentage of oxides, generally Al2O3, in order to achieve a homogeneous dispersion of oxide nanoparticles in the metal matrix. The items manufactured after consolidation of the milled powders had an improved strength that was maintained up to near 90% of the melting point and they were termed oxide dispersion strengthened (ODS) alloys. Excellent reviews on the synthesis of ODS alloys have been previously reported in the literature.14,23,24,496 It must be pointed out that the milling of a ductile metal leads to a dramatic growth of the particles because of agglomeration by cold welding. Thus, they cannot be mechanically alloyed unless a process control agent (PCA) was previously added to prevent cold welding. More generally, used PCAs have been reviewed in literature.24
It must be remarked that innovative techniques like spark plasma sintering (SPS), hot isostatic pressure (HIP) or hot extrusion (HE) have been developed to achieve a full-density compaction at temperatures considerably lower than the melting point, which would allow the metastable phases and nanostructures developed by milling to be retained.24,496 This situation has dramatically enhanced the interest in developing new ODS aluminum, steel or ferrous superalloys and metal/ceramics composites either using alumina497–503 or other ceramic-dispersed phases like TiC,504–509 TiB,510,511 BN,512 carbon nanotube,513,514 fullerene,515 ZrO2,516 TaC,517 NbC,518 TiN,519 Cr2O3,520 SiC521 or WC522,523 with a noticeable improvement of the mechanical properties.
The synthesis of Cu-dispersed Al2O3 nanocomposites by high-energy ball milling and pulsed electric current sintering was studied by Kim et al.524 The nanocomposite powders with crystallite size of about 25 nm were successfully synthesized by a high-energy ball milling process. The composites sintered at 1250 °C for 5 min showed a relative density above 97% and enhanced fracture toughness of 4.51 MPa m−2. Microstructural observation of the sintered composites revealed that the nanosized Cu particles were uniformly distributed and situated on the grain boundaries of Al2O3 matrix. Dense nanocrystalline TiB2–TiC composites have been formed by field activation of high-energy ball-milled reactants.525 Elemental powders (Ti, C, B) were milled in a planetary mill to produce nanometric powders without product formation. No significant amount of any of the two product phases was found to be formed until 3 hours of milling. The as-milled powders reacted under the influence of high current (ac current 1800 A) and uniaxial pressure (30 MPa applied for 3 min). Dense (up to 98.6%) nanocomposites were formed. The crystallite sizes of TiB2 and TiC in the dense composite formed from powders milled for 10 hours were 71.4 and 62.5 nm, respectively. The microhardness of this composite was 20.6 GPa. El-Eskandarany et al.526 have succeeded in the synthesis of nanocomposite WC/MgO powders by mechanical solid-state reaction and subsequent plasma-activated sintering. A mixture of elemental Mg, WO3, and C powders was milled under argon gas atmosphere at room temperature using high-energy ball milling. Nanocomposite powder WC/MgO (17.6 at% MgO) was obtained after milling. Plasma-activated sintering was applied to consolidate this end product into a fully dense bulk sample. The consolidation step does not lead to extensive grain growth and the compact sample consists of nanoscale grains of MgO (2.5 nm in diameter) embedded into the matrix of WC. This as-fabricated nanocomposite has high values of hardness (about 15 GPa) and fracture toughness (14 MPa m−2). In order to produce alumina–Cr and alumina–Nb composites, the mixtures of Cr–Al2O3 and Nb–Al2O3, respectively, were milled in attritor in the equivalent volumes (50 vol% Al2O3 and 50 vol% Cr or Nb). The obtained composites have been densified from 1310 to 1450 °C in about 10 min. The applied pressure was 45 MPa.527 The milling intensity applied on the metal–Al2O3 powders has been found to play a fundamental role determining not only the degree of densification, but also the final nature of the composites. Longer milling time led to material exhibiting finer microstructure. The maximum fracture strength developed in these composites has been achieved by sintering at 1380 °C, reaching 825 MPa for the Nb–Al2O3 materials. Murakami et al.528 studied the microstructure, mechanical properties and oxidation behavior of powder compacts of the Nb–Si–B system prepared by spark-plasma sintering. The study was performed on sintered compacts with eight different compositions in the Nb5Si3–NbB2–NbSi2 triangle. Compacts were prepared via three different routes: (i) blending elemental powders in rotational ball mill and consecutive sintering, (ii) mechanical alloying of elemental powders and consecutive sintering, and (iii) presintering blended elemental powders, consecutive milling and finally sintering them. The most homogeneous and the least oxygen-contaminated compacts were obtained via the third route.
6. Peculiarities of mechanochemical reactions
6.1 Soft mechanochemical reaction
Avvakumov et al. elaborated an original method referred to as soft mechanochemical synthesis for mechanochemical reactions with the participation of highly reactive compounds.16,529–532 The soft mechanochemical processes mean that highly reactive functional groups are involved in the reactions the –OH group is by far the most important.533 It was also shown that the reaction rate with close values of reaction heat is controlled by the hardness of reagents: the lower the hardness, i.e. the higher the plasticity of the substance, the closer the contact between the particles and the higher the reaction rate.532 Much lower energy is required for these mechanochemical reactions in comparison with conventional dry or anhydrous ones.16,530,533 Products of soft MCS are quite often either in a completely non-crystalline or severely disturbed crystalline state. One of the most promising prospects for soft MCS is the preparation of desired crystalline compounds without further heat treatment.530,534 Another benefit of these processes is an appreciably lower temperature necessary to obtain well-crystallized final products. This is advantageous not only because of the energy saving, but much more importantly, because of having smaller crystallites or grains without suffering from an excessive or abnormal grain growth.534Phase purity, high crystallinity and small grains with narrow size distribution are almost always required for most of the functional composite oxides. Two factors are of utmost importance, i.e. (i) high number of nucleation sites and (ii) short diffusion path for subsequent growth. They also favor kinetic stabilization, which is indispensable to avoid falling into thermodynamically more stable phases in the course of cooling. Soft mechanochemical synthesis,16 by mechanically activating the starting mixture, is one of the best options to the goal. The main spirit is the reconstruction of the electronic states of the mixture closer to the final products, by virtue of charge transfer at the contact points of dissimilar solid species contained in the reaction mixture.535 An acid–base reaction at the interface between acidic oxide and basic hydroxide by co-milling SiO2 or TiO2 with Ca(OH)2, Sr(OH)2 or Al(OH)3 is the central point of the soft mechanochemical processes,536–539 while mechanisms via radical recombination could not be ruled out.540
The sol–gel route is generally out of the scope of mechanochemistry. However, it is important to note that atomic-level homogenization is possible by milling a mixed gel. This was demonstrated especially in detail for lead titanate.541,542
Co-milling a stoichiometric mixture of (hydr)oxides results in highly reactive precursors of mixed oxides. Several examples will be given below. Phase-pure ilmenite MgTiO3 with its quality factor (Q) as high as 9800 was obtained from a co-milled stoichiometric mixture of Mg(OH)2 and TiO2.543 A solid solution, 0.9Pb(Mg1/3Nb2/3)O3 × 0.1PbTiO3 (0.9 PMN × 0.1 PT) was obtained in a pure perovskite phase by starting from a co-milled stoichiometric mixture of PbO, TiO2, Mg(OH)2 and Nb2O5 by calcining at 850 °C for 4 h.544 The maximum dielectric constant of 24600 was attained after sintering at 1200 °C. A similar technique was applied to the more challenging system of a PMN-PZN solid solution to obtain phase-pure perovskite as well.545
6.2 Acid–base reactions
Different approaches are used to explain the acid–base properties of solid substances. These approaches are based mainly on the Brønsted–Lowry and Lewis acid–base definitions. Valuable information of the acid–base properties of many solid substances can be found in ref. 16. One of the most important advantages of acid–base processes is the utilization of chemical spontaneity or reactivity during ball milling to the maximum extent. This valuable concept can be used for the synthesis of many compounds where an acid–base interaction can be expected. However, in some cases, mainly when anhydrous starting compounds have been used, acid–base synthesis was not successful even if long-time mechanical milling has been performed.532,546,547 An explanation of such discrepancies relies on a kinetic scheme. A Brønsted–Lowry acid–base reaction in the presence of –OH groups or hydrated acidic compounds such as metal chlorides, nitrates, sulphates, etc., is followed by a fast proton transfer with low activation energy, accompanied by a neutralization reaction. On the other hand, a Lewis acid–base reaction (ligand exchange or displacement reaction) in –OH-free or H2O-free conditions caused complicated solid-state ligand exchange reactions and the cleavage of covalent bonds connected with much higher activation energy. In such cases, the subsequent annealing is needed to complete the final reaction.17,546,547 Remarkably, by selecting suitable conditions such as chemical reaction paths, stoichiometry of starting materials and milling conditions, acid–base MCS can be used to synthesize nanoparticles dispersed within a water-soluble salt matrix,548 where the new-formed salt matrix represents an analogy for salt formation via the traditional neutralization process. Nanoparticles formed as two-dimensional nuclei at points of contact between reagents are dispersed in the salt matrix, which is just like the plums within the pudding in Thomson’s ‘plum pudding’ model of an atom. The nanoparticles then grow in volume.532,549 The resulting nanoparticles show low agglomeration and uniformity of crystal structure and morphology, which are unique features of MCS nanoparticles and are superior to the nanoparticles produced by other techniques.17,548Fig. 30 schematically illustrates the acid–base strategy resulting in the formation of oxide nanoparticles. Fig. 30a shows the starting microcrystalline acid–base mixture. The constant formation of salt matrix shells during milling surrounds the newly formed nanoparticles and prevents them from aggregating (Fig. 30b). The crystallinity of nanoparticles can be further improved by heat treatment (Fig. 30c) prior to the removal of the solid salt matrix phase by a simple washing procedure (Fig. 30d). 1D morphology of oxide nanoparticles prepared by molten salt treatment is shown in Fig. 30e and oxide nanoparticles obtained after washing with water are shown in Fig. 30f.17,549–552
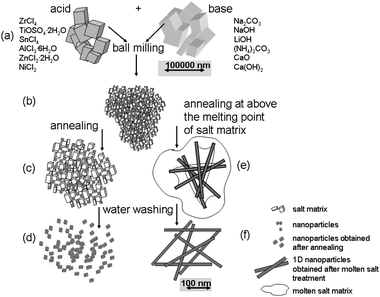 | ||
| Fig. 30 Mechanochemical synthesis (MCS) of oxide nanoparticles via an acid–base strategy. | ||
Fig. 31 shows TEM images of various oxide nanoparticles prepared via MCS. Further details related to the preparation procedure can be found in ref. 17.
 | ||
| Fig. 31 TEM images of oxide nanoparticles prepared via MCS: (a) TiO2, (b) α-Mn2O3, (c) Na2Ti6O13, (d) SnO2, (e) VO2, (f) α-Al2O3. | ||
Table 4 shows examples concerned with the synthesis of nanocrystalline oxides. In some cases, NaCl, LiCl or Na2SO4 have been used as a diluents because of their inert nature and excellent milling.547,551,553 Moreover, the addition of an inert salt diluent into the starting powder prevents the occurrence of agglomeration or combustion because: (1) the diluent separates the reactants; (2) the diluent absorbs some of the collision energy during milling, reducing the energy transfer into the reactants; (3) the diluent absorbs heat generated by the reaction and reduces the temperature reached during milling. Unfortunately, in some cases, the dilution of reactants with NaCl substantially hinders the mechanochemical reaction.551
Table 4 Examples of chemical strategies of oxide nanocrystals production by acid–base mechanochemical synthesis. Further details can be found in ref. 17
| Compound | Reaction scheme or starting compositions | Milling time, post-milling conditionsa,b, crystallite size calculated from XRD or TEMc |
|---|---|---|
| a N – the desirable reaction product was not observed directly after milling.b Lowest annealing temperature necessary to obtain the desirable nanocrystalline product.c Mean crystallite size determined from the XRD line broadening by using the Scherrer equation. | ||
| ZrO2553 | ZrCl4 + 4LiOH → ZrO2 + 4LiCl | 1 min, —, 9 nm |
| CeO2550 | 2CeCl3·6H2O + 3Na2CO310H2O | 5 min, 900 °C, 40–70 nm |
| SnO2555 | SnCl2 + Ca(OH)2, K2CO3 → SnO + CaCl2, 2KCl + CO2 | 3 h, 400 °C, 11 nm |
| TiO2556 | TiOSO4·2H2O + Na2CO3 → TiO2 + Na2SO4 + CO2 + xH2O | 5 min, 400 °C, 8 nm |
| Al2O3546 | 2AlCl3 + 3CaO → Al2O3 + 3CaCl2 | 24 h, N, 1250 °C, 50–100 nm (TEM) |
| Fe2O3557 | FeCl3·6H2O + 6 NaOH → Fe2O3 + 6 NaCl + 9H2O | 30 min, —, 15 nm (TEM) |
| NiO547 | NiCl2 + Na2CO3 + 4NaCl (as diluent) → NiO + 6NaCl + CO2 | 6 h, N, 400 °C, 10 nm |
In contrast to the traditional isolation of the as-prepared nanocrystalline powders where a washing procedure is always needed after annealing, a new procedure combining the crystallization of oxide powders with the evaporation of the salt matrix was introduced.17,552,554 The idea is based on the physical properties of newly formed NH4Cl salt matrix instead of NaCl, LiCl or Na2SO4, because NH4Cl decomposes or sublimates during the following low-temperature annealing. Furthermore, by utilization of this method it is possible to avoid contact among nanocrystalline oxide materials and washing water, which can lead to undesirable reactions.17 TiO2 and SnO2 nanoparticles (Fig. 31d) have been prepared by this unique strategy.17,552,554
6.3 Solid-molten salt reactions
A procedure has been developed in which the MCS or MA is combined with a subsequent treatment in a molten salt matrix.17,558–562 The aim is to provide enough activation energy to the pre-activated sample so that the chemical reaction is completed in the following molten salt synthesis. Unlike the traditional MCS of nanoparticles in a salt matrix, solid-molten salt synthesis is based on annealing at a temperature above the melting point of the newly formed salt matrix, or with other words in the molten salt. Nanosized carbides such as TiC, Mo2C or WC have been successfully synthesized by this method.558–560 The role of the molten salts is: (1) to increase the reaction rate and lower the reaction temperature; (2) to control particle shapes; (3) to control particle sizes; (4) to increase the degree of homogeneity; (5) to control the agglomeration state.563Typical examples of salts used in molten salt synthesis are chlorides (NaCl) and sulphates (Na2SO4). In many cases, eutectic mixtures of salts are used to lower the liquid formation temperature.563 Likewise to the case of the acid–base MCS of nanopowders, the salt matrix is eliminated after cooling by washing with water (Fig. 30f). Furthermore, the role of the molten salt can be extended effectively and powders with unique 1D (Fig. 30e) or 2D morphology can be easily prepared.17 For example, Na2Ti6O13 phase with 1D belt-like morphology has been obtained from a nanocrystalline TiO2–NaCl–Na2CO3–Na2SO4 mixture after annealing at 800 °C (Fig. 31c).17,561 MA of the Fe2(SO4)3–Na2CO3–NaCl mixture followed by annealing at the temperature close to the eutectic temperature of the Na2SO4–NaCl salt matrix leads to 2D Fe2O3 platelets.562 In this context, low-temperature mechanochemical molten salt synthesis of 2D α-Al2O3 nanoplatelets by using reactive Al2O3–NaCl–KCl mixture has been verified.17
It is believed that the features of the molten salts are related to the surface and interface energies between the nanocrystalline solid constituents and the salt liquid, resulting in the tendency to minimize the energies by forming a specific morphology. Therefore, the selection of salt is critical in obtaining desirable powder characteristics.563
6.4 Redox reactions
In general, these reactions can be expressed by equation:| Me1X + Me2 → Me1 + Me2X | (10) |
where Me1 is reduced metal, Me2 is reducing metal, X is oxygen, chlorine, fluorine, sulphur, etc. Solid-state redox reactions involve simultaneous oxidation and reduction processes. According to present views, a solid-state redox reaction is a reaction in which a transfer of electrons from reducing metal to reduced metal takes place. Sometimes they are called solid-state exchange or metathesis reactions (SSM).564
These are solvent-free reactions that enable a wide range of ceramic materials to be made in seconds. These fast solid-state reactions take advantage of the reaction enthalpy released in a specific reaction. Typically, in SSM a salt is formed along with the desired product. The high-lattice energy of the co-formed salt provides the driving force of the process.565
Mechanochemical solid-state reduction of oxides can be schematically expressed by general equation:
| Me1O + Me2 → Me1 + Me2O | (11) |
where Me1 is reduced metal, Me2 is reducing metal. A large variety of Me1 and Me2 combinations has been used, e.g. Me1 = Fe, V, Cr, Mn, Co, Ni, Ti, Sn, Pb, Cu, Zn, Nb, Mo, W, Si and Me2 = Al, C, Ti, B, Cr, Si, Mg, Zr, Ca, Zn, Ni and Mn.7 The reaction is thermodynamically feasible, if the heat of formation (per oxygen atom) is larger for Me1O than for Me2O.
To test whether this concept can be used as a vehicle for the chemical reduction of oxides, the powders of copper oxide and carbon were milled.566
| CuO + C → 2Cu + CO2 | (12) |
This reaction is thermodynamically feasible at room temperature (ΔH = −138 kJ mol−1), but will not occur for kinetic reasons. The reduction was found to occur via a two-stage process (CuO → Cu2O → Cu). The effect of milling on the phase present was determined from quantitative analysis of XRD patterns and it is shown in Fig. 32. Examination of the powder using transmission electron microscopy (TEM) showed the as-milled particles contained small Cu crystallites of 15–30 nm in size.
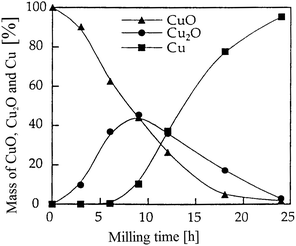 | ||
| Fig. 32 Effect of milling time on the two-stage reduction of CuO by carbon.566 | ||
Mechanochemical reduction is an extension of the mechanical alloying process (see part 5.2). In these highly exothermic reactions, it has been shown that mechanical alloying can cause a reaction which proceeds by the propagation of a combustion wave through the partly reacted powders. The combustion conditions are reached after a critical ignition time (tig), which has been shown to be the milling time required for the ignition temperature to be reached (Tig).566 The ignition and propagation of self-sustaining reactions is governed by the principles of conventional SHS,25 see part 6.5. The first SHS solid-state reaction was proposed by Goldschmidt in 1885.567 It is the so-called thermite reaction in which a metal oxide is reduced with aluminum. The process takes place in a mixture of powders and is initiated by either high temperature or an electrically heated wire. A combustion front develops and propagates across the sample.25
Sulphides. Sulphides play an important role in traditional technological applications as well as in the field of advanced materials. Solid-state reactions of sulphides to prepare the elemental nanometals or their oxides proceed via two routes:
| Me1S + Me2 → Me1 + Me2S | (13) |
| Me1S + CaO → Me1O + CaS | (14) |
In redox reaction (13) the reduction of the metal sulphide Me1S is performed with a reducing element Me2 (usually Fe, Mg, Al, Si). Reaction (14) represents a displacement reaction. Sometimes a combined effect of redox and displacement reaction is applied.
The mechanochemical reaction between copper sulphide and elemental iron has been studied very carefully.568–572 The reaction can be described by equation:
| Cu2S + Fe → 2Cu + FeS | (15) |
Reaction (15) is thermodynamically feasible, as the enthalpy change is negative (ΔH°298 = −21.0 kJ mol−1). The progress of the mechanochemical reaction is illustrated by the selected XRD patterns in Fig. 33. The primary process (the reduction of copper sulphide by iron while copper metal and iron sulphides are formed) is clearly seen, particularly by inspecting the relative intensities of the diffraction lines of Cu and Fe metal.
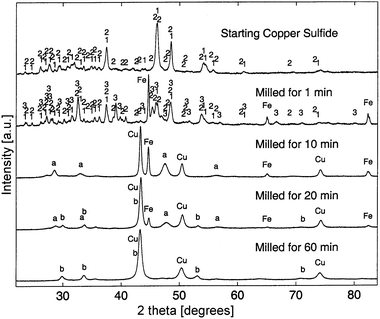 | ||
| Fig. 33 X-ray diffraction patterns for reaction (15) as a function of milling time, 1 – djurleite Cu1.94S, 2 – chalcocite Cu2S , 3 – tetragonal Cu1.81S, a – cubic FeS, b – hexagonal FeS. Reprinted with permission from ref. 571. Copyright 2002, Trans Tech Publications. | ||
Most of the copper sulphide is consumed during the first minutes of milling. After 10 min, only the reflections of Cu, cubic FeS (JCPDS 23-1123) and Fe can be observed. The presence of iron without residual copper sulphides requires either significant non-stoichiometry of the iron sulphide or the presence of free sulphur. Unfortunately, free sulphur may remain undetected by XRD due to the low scattering amplitude of sulphur and its tendency toward amorphization. The intensity of the iron reflections decreases upon continued milling and no iron is detectable after 60 min. Simultaneously, FeS is changed to its hexagonal modification (troilite, JCPDS 75-0602).
Micrometer aggregates of nanoparticles are formed during milling of copper sulphide with iron (Fig. 34). Usually each micrometer-sized powder particle is comprised of a mixture of nanosized grains of the product phases, as can be observed in our case.573–575 The dimension of the grains changes with milling time (Fig. 34).
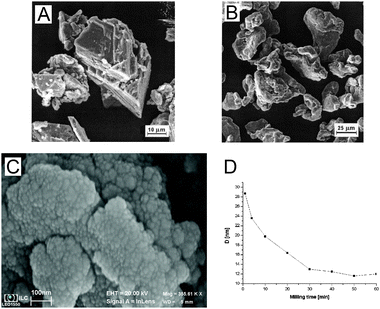 | ||
| Fig. 34 SEM images of precursors for reaction (15): A – copper sulphide; B – elemental iron; C – TEM image of produced Cu/FeS; D – dependence of crystallite size D on product copper for reaction (15) as a function of milling time. Reprinted with permission from ref. 569. Copyright 2005, Interscience Publishers. | ||
Mechanical milling of ZnCl2 and CaS mixtures resulted in the formation of 500 nm ZnS particles containing aggregates of 12 nm crystallites according to the following reactions:576
| ZnCl2 + CaS → ZnS + CaCl2 | (16) |
| ZnCl2 + CaS + 3.6CaCl2 → ZnS + 4.6CaCl2 | (17) |
| ZnCl2 + CaS + 8.2CaCl2 → ZnS + 9.2CaCl2 | (18) |
The addition of 71 vol% CaCl2 as a diluent to the reactants resulted in the formation of separated 16 nm particles of ZnS. Using mechanically alloyed CaS (10–50 nm particle size) enabled the synthesis of isolated ZnS particles with size of 7–9 nm.
Solid-state room-temperature redox reactions of metal sulphides (FeS, WS2, MoS2, CoS, PbS, ZnS)577 were conducted by ball milling of powder mixtures of sulphide and reductant (R = Al, Mn, Fe, Si) or an exchange compound (CaO). Milling results in a complete reduction of the metal sulphide and in the formation of nanocomposites composed of elemental metal and R sulphide. Transformations follow eqn (13) and (14). Exchange reactions with CaO show the formation of M oxide–CaS nanocomposites.
Iron sulphides were, for instance, reduced by aluminum, manganese and silicon; copper sulphides by aluminum and cobalt; lead and zinc sulphides by aluminum. Redox reactions were performed in the cases of iron, tungsten, and molybdenum sulphides. The preparation of MoS2 nanoflakes has also been obtained by mechanochemical synthesis.578
Bi2S3 polycrystals were also synthesized by a simple process combining mechanical alloying with spark plasma sintering technique.579 The electrical resistivity was significantly reduced by modification of sulphur content through producing sulphur vacancies and by increasing the carrier mobility through forming oriented microstructure from 91 mW m K−2 for Bi2S3 to 181 mW m K−2 for Bi2S2.90, where the power factor was significantly enhanced by modifying sulphur contents, and further increased to 254 mW m K−2 for the textured Bi2S2.90 obtained by hot forging. The thermal conductivity of Bi2S3 ranging from 0.83 to 0.89 W m K−2, which is comparable to or even lower than that of Bi2Te3 compounds was observed. An enhanced maximum ZT value of 0.11 at 523 K was achieved for the textured Bi2S2.90 sample, which is the highest value known presently in the Bi2S3 system. Further enhancement of electrical transport properties could be accomplished by optimizing the carrier concentrations and mobilities through accurately controlling the stoichiometry and grain orientation. Bi2−xAg3xS3 (x = 0–0.06) polycrystals were fabricated by mechanical alloying and spark plasma sintering.580 The phase, microstructure, electrical and thermal transport properties were investigated with special emphasis on the influence of Ag doping content. All the Bi2−xAg3xS3 powders can be indexed as a single phase with an orthorhombic symmetry. Second phase AgBi3S5, which occurs at doping content x ≥ 0.02, enhances the electrical conductivity. This results in the improvement of electrical transport properties of Bi2−xAg3xS3 bulk samples. A maximum ZT value reached 0.23 at 573 K for the Bi1.99Ag0.03S3 sample, which is 130% higher than in case of the pure Bi2S3.
The suitability of silver sulphide (Ag2S) and its nanocomposite Ag2S/C, prepared by simple solid-state synthetic routes such as high-energy milling and heat treatment, has been investigated.581 This material can be utilized as anode material in rechargeable Li-ion batteries. Well-developed Ag2S phase can be identified from X-ray diffraction patterns, and XRD and HRTEM observations confirm that Ag2S nanocrystallites in Ag2S/C nanocomposite are uniformly distributed within an amorphous carbon matrix. The mechanism of electrochemical reaction between Ag2S and Li is identified by ex situ XRD analyses of an Ag2S/C nanocomposite electrode. The Ag2S/C nanocomposite anode shows good cycling behavior and high capacity of about 430 mA h g−1 after 100 cycles.
6.5 Self-sustaining reactions
Mechanically induced self-sustaining reactions. Mechanochemistry is generally performed in high-energy ball mills using powder reactant mixtures. During milling, the intimate mixing of reactants and the continuous creation of fresh surfaces, defects and active sites enable the gradual progression of solid state reactions at room temperature by small diffusion paths at the interfaces of the reactants due to the ball impacts. If these solid-state reactions are highly exothermic, a self-sustaining reaction similar to thermally ignited self-propagating high-temperature synthesis (SHS) can be initiated within the vial after a critical milling time called the ignition time (tig) and the majority of the reactants are consumed within seconds. This type of mechanochemical process is referred to as a mechanically induced self-sustaining reaction (MSR)25 and has been reported in the synthesis of chalcogenides,582 carbides,377 nitrides,406 borides,391 silicides,439 aluminides583,584a and metals.585
The self-heating capacity of mixtures submitted to milling determines the appearance of the MSR process. This capacity is associated with the adiabatic temperature (Tad), which is defined as the maximum temperature reached by the products due to the reaction heat if it occurs in an isolated system. SHS reactions are empirically suggested not to occur unless the adiabatic temperature is greater than 1800 K.586 In principle, the same rule could be applied to MSR processes. However, it has been observed that not every SHS reaction can be initiated by ball milling. In MSR, the reaction powder mixture is not compact and is in contact with the milling media. A significant amount of heat is released to the environment, which can prevent the formation of a propagating front. Consequently, only extremely high exothermic systems exhibit the MSR effect. High Tad is an indispensable requirement to produce MSR, but other experimental parameters should also be controlled to guarantee the combustion process. Indeed, a literature review reveals that there is not always a straightforward relation between Tad and tig. A more exothermic reaction does not always ignite after a shorter milling time.25,391 The tig strongly depends on the milling intensity, although the thermomechanical properties of the reactant mixture also play an important role. tig values of seconds, minutes and even hours have been observed.391
For a diffusion-controlled mechanochemical reaction, the conversion as a function of the milling time is generally characterised by a sigmoidal curve with three different stages.587 First, there is an incubation time during which no product is formed that corresponds (as for any other alloying process) with the mechanical activation of the powder. Comminution, mixing and defect formation take place during this period. After this incubation time, when the reactant mixture is sufficiently activated and reactive, the subsequent ball impacts induce the onset of the mechanochemical reaction, and an increase in the conversion rate (acceleratory stage) is observed. Finally, as the availability of fresh reactant surfaces decreases, the reaction rate slowly diminishes with prolonged milling (deacceleratory stage) until the complete conversion is reached.
In a highly exothermic system, after the incubation time, the mechanochemical reaction is initiated due to ball impacts, but the sufficient heat to initiate and multiply the ignition points required to spread out the self-sustaining reaction can be released. Occasionally, the incubation and ignition times practically coincide and therefore no product is observed before ignition.385,435 However, in other cases, the conditions for ignition are achieved when the mechanochemical reaction has already partially progressed by a diffusional mechanism, and the products are clearly detected before ignition.403,407 It is also possible that before reaching these optimal conditions for ignition the product formed by diffusion inhibits ignition similarly to an inert additive. If the self-sustaining reaction does not begin before the formation of a threshold amount of product, the MSR process cannot occur, and the reaction will gradually progress. This process explains why both gradual and combustive kinetics are possible for the same system depending on the milling conditions.
In comparison with other mechanochemical processes, MSR has the advantage of tig. This reproducible parameter can be measured by detecting the abrupt temperature increase of the milling vial583 or the total pressure inside the vial455 as a consequence of the heat released from the highly exothermic reaction. Some studies have focused on systematic investigations of the effects of experimental milling conditions on the ignition of self-sustaining reactant mixtures. Using the reduction of CuO with Fe, Schaffer and McCormick588 observed an inverse relationship between the ball-to-powder ratio (BPR) and tig. Schaffer and Forrester589 examined the effects of the impact energy on the formation of TiC by varying the ball density while keeping the ball diameter and the BPR constant, and they also observed a similar inverse relationship between BPR and tig, but tig was also observed to be independent of the ball mass and, therefore, they concluded that increasing the collision energy does not always decrease tig. However, Deidda et al.590 observed strong dependence of tig on the impact energy during the synthesis of TiC that was varied by modifying the frequency of the mill. Kudaka et al.591 studied the effects of zirconia, steel and WC milling media on the mechanochemical synthesis of Ti5Si3 using the same BPR and rotation speed of a planetary mill. The authors observed the shortest tig values for zirconia, the low-density milling medium, which was attributed to a lower thermal conductivity that allowed the heat produced during milling to be effectively stored.
These contradictory results were in part the consequence of using BPR as the primary parameter to characterise the intensity of the milling processes, while neglecting the dynamics of milling that determines both the collision frequency and the impact velocity. To maintain a constant BPR when the ball density is altered implies that the number of balls in the vial is changed and, subsequently, the number of impacts per unit time, or the quantity of powder and then the mechanical dose rate (energy transferred to the powder per unit mass and time). Deidda et al.590 reported the existence of an impact energy threshold below which ignition does not occur. However, if this threshold is exceeded it appears that the mechanical dose required to ignite a specific reactant mixture is approximately constant independent of the impact energy. If milling is performed at different milling intensities, ignition occurs at different time scales.592
Because tig can be used as a reference point due to its variation with processing conditions that reflect changes in the mechanical dose rate of the mill, this parameter also facilitates the comparison of reaction kinetics data obtained using different milling equipments and consequently their efficiencies.593
Mechanically activated self-heat-sustaining reactions. SHS (self-heat sustaining reactions) provide a suitable method for producing advanced materials, such as ceramics, composites and intermetallics. The SHS process offers advantages with respect to process economy and simplicity. The basis of such a synthetic method relies on the ability of highly exothermic reactions to be self-sustaining and therefore energetically efficient. If a very exothermic reaction between solids or solid and liquid reactants is locally initiated, it may generate enough heat to ensure the propagation of a transformation front leading to an end product involving all the initial amounts of the elemental components. Such a process is characterized by a fast moving combustion front (1–100 mm s−1) and a self-generating heat which leads to a sharp increase of temperature, sometimes up to several thousands of K s−1. The temperature reached inside the reaction front has been found to be effective to volatilize low boiling point impurities, helping by the way to produce purer end products than those obtained by some more conventional techniques. Furthermore, if the temperature variation after the end product formation is kept under control, no chemical macrosegregation occurs.
The new version of the SHS process introducing mechanical activation as a first step (before the ignition of SHS reaction) – so-called MASHS (mechanically activated self-heat sustaining reactions) was proposed by Gaffet in 1995 for the synthesis of the nanocrystalline FeAl compound.584
Such a MASHS process has been successfully used to obtain various nanocrystalline compounds584b,c such as FeAl,594–598 MoSi2,599,600 FeSi2,601,602 Cu3Si,603–605 NbAl3606–609 and Cu3Si.604,609b The effect of mechanical activation on the combustion synthesis of NbAl3 has been more recently investigated by Neto and Da Rocha.610 As milling time is increased, the ignition temperature is decreased to temperatures below the melting point of aluminum, going from 850 to 500 °C. Also a variation in the reaction pathway is observed, changing from two-stage to single-stage. Uenishi et al.611 have succeeded in the synthesis of nanostructured titanium aluminides and their composites formed by combustion synthesis of mechanically alloyed powders. Application of mechanical activation and TiB2 addition was confirmed to be effective for the suppression of grain growth as well as for the nanostructure synthesis caused by the subsequent heat treatment, but it made the sintering conditions more difficult when full densification has to be achieved. Such a combustion synthesis of ball-milled powders has also been applied to the fabrication of a thick layer of the intermetallic compound Al3Ti on a metallic substrate.612 A thick layer of Al3Ti was formed on a Cu or Ti substrate by a coating process involving combustion synthesis of a ball-milled Al–Ti powder mixture. This layer has almost the same wear property and hardness as that of cast Al3Ti.
The influence of mechanical activation on the self-propagating high-temperature synthesis has been investigated on reactions that are difficult to initiate, e.g. Ni3Si formation.613 Self-propagating high-temperature synthesis of Ni3Si is difficult, if not totally impossible, without mechanical activation.
The combustion synthesis of α-AlFeSi intermetallic phase has been investigated by Murali et al.614 The speed of ball milling is an important parameter in the combustion synthesis of intermetallics from elemental powders.
In order to control this type of reaction, a better understanding of the initial conditions controlling the ignition step is requested, since the initial conditions are the easiest to control, if not the only parameters allowing the control of the reaction. Many different characterization techniques have been used in order to obtain such information.615 Until recently, it has been difficult to investigate these reactions by conventional techniques due to the high temperatures and high rates of combustion. Conventional techniques do not permit the study of the intimate mechanisms of such exothermic reactions, e.g. the role of liquid formation and the existence of a transitory phase and other parameters which may induce changes of the texture or nature of the end product. Recently, real-time in situ investigations of structural changes and chemical dynamics in the combustion area have been made possible by the use of synchrotron radiation.616–618 Specific attention has been paid to the MASHS process applied to the synthesis of NbAl3,619 MoSi2620–622 and FeAl.623
6.6 Role of water in mechanochemistry
Water is a polar liquid which shows extraordinary properties attributed to the presence of hydrogen bonds. The structure of water and the explanation of its properties continues to be a subject of major debate.624 As early as 1932, Alexander and Byers625 observed the evolution of hydrogen during the milling of wet sand in a vibration mill. Decomposition of water by sound waves is experimentally confirmed.626 Clusters of water molecules present in liquid water can break under mechanical influence and reshape after its termination. Mechanical motion of pure water can also lead to localised changes in electrical potential.627 Styrkas and Nikishina627 have recently reviewed the mechanochemical processes in water. However, the focus here is on solid–water systems under a mechanochemical environment. The properties of water at the interface between liquid and solid phases differ from its bulk properties. Principles governing the mineral/water and particle/water interfaces apply to mechanochemical phenomena.628,629 Water is encountered in mechanochemical reactions in various forms. Some of these are depicted in Fig. 35. Few publications deal with the role of water in mechanochemistry.629–634 In this part we provide a critical update and touch upon the genesis of water in mechanochemical reactions, associated phenomena/effects and select examples to highlight various roles of water. Hydrothermal aspects633 and wet stirred high-energy ball milling634 are especially highlighted.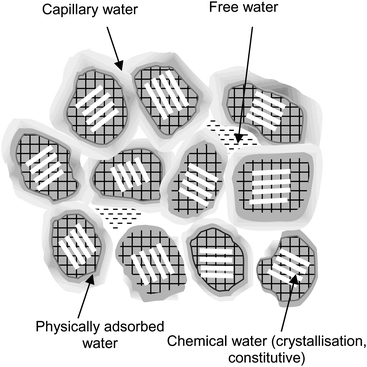 | ||
| Fig. 35 Various forms of water encountered during mechanochemical reactions. | ||
Genesis of water in mechanochemical reactions. The origin of water in mechanochemical reactions may lie in the solid phase(s) or it may be water which is extraneously added. Crystallized water (as in CuSO4·5H2O, FeSO4·7H2O, CaSO4·0.5H2O) and constitutional water formed by ions of OH− and H+ lost by minerals (for example, hydroxides and oxyhydroxides, e.g. M(OH)n, MO(OH)n (where n is the valency of metal (M) ion such as Al3+, Ca2+ and Mg2+); clays like kaolinite (Al2(Si2O5)(OH)4); etc.) may be liberated/formed and squeezed out due to mechanical milling.630 Water may also be formed during solid–solid reactions, e.g. soft-mechanochemical reactions between hydrated oxides.631 Extraneous water may be present in various forms from vapour to solid–liquid pastes (as in kneading or pug milling635,636) and slurries (wet milling634). It is important to appreciate the dynamic nature of water and its effect in mechanochemical treatment. Initially, the solids containing crystallised water may be dry and as the milling progresses water is liberated and it may be present in the pores, can spread out as physically/chemically adsorbed water, may occupy interparticle space or may be present as capillary water depending on the duration of milling and system under consideration. Some solids may be inherently porous, e.g. aerosilogel and silica gel,637 and may contain water in their pores. The wet milling may influence particle–particle interaction and rheology of the solid–water suspension.638 The energy transfer from media to material and its mechanism is dependent on the quantity of water present.633 During milling, water also serves as dissipation media for milling energy. Fig. 35 presents a very simplified picture of various forms of water which can possibly occur during mechanochemical reactions. There are numerous interactions possible during milling among different entities including solids, water, milling media and milling chamber and these interactions can be influenced by the milling environment. The overall effect of water is governed by the nature of solid (porous and non-porous, crystalline, microcrystalline, amorphous, hydrous or anhydrous, etc.), genesis and quantity of water, the type of mill, milling mechanism and milling conditions (milling time, milling intensity, etc.).
Mechanochemical effects in the presence of water. The presence of water during mechanochemical treatment may influence particle breakage as well as the mechanical activation of individual solids and their reactions.629–634 The enhanced breakage and milling efficiency may result from the lowering of solid strength due to decrease in interfacial tension (Rebinder effect), increased mobility and annihilation of dislocation and/or surface charge (chemo-mechanical effect) and retardation of sealing of freshly created cracks.629,630,639–642 The phenomena of aggregation and agglomeration (see section 1.4) responsible for coarsening of size distribution during milling and mechanical activation are affected by the presence of water.629,630,634 In the presence of a polar liquid like water, molecular dense aggregates decompose to give primary particles and fine milling rather than mechanical activation is promoted.629,630,634 However, if the water forms 1–3 layers on the surface of the particles, it acts as a binding agent and improves the strength of the agglomerates through the formation of hydrogen and coordination bonds between water molecules and a hydroxylated (![[double bond, length as m-dash]](./Hallmarks_files/char_e001.gif) Si–OH, Al–OH, etc.) surface. Due to the structuring of water layers and the action of the surface field on water layers, hydrogen bonds become stronger; their energy becomes higher by a factor of 2–2.5 than that of hydrogen bonds in the bulk water and water itself exhibits different properties (ρ = 1.2 g m−2 and dielectric permeability ε ≤ 5).631 Agglomeration can be attributed to capillary pressure and/or chemihesion (the presence of chemical bonds exactly at the places of contact among solids). The overall stability of the dispersed system may be influenced by the interplay of several phenomena occurring simultaneously, e.g. adhesion, chemihesion, capillary pressure, etc.629,630,634 Amorphisation is often accompanied by a condensation process within the crystal lattice. This is typical for kaolinite and it is shown in Fig. 36.629
Si–OH, Al–OH, etc.) surface. Due to the structuring of water layers and the action of the surface field on water layers, hydrogen bonds become stronger; their energy becomes higher by a factor of 2–2.5 than that of hydrogen bonds in the bulk water and water itself exhibits different properties (ρ = 1.2 g m−2 and dielectric permeability ε ≤ 5).631 Agglomeration can be attributed to capillary pressure and/or chemihesion (the presence of chemical bonds exactly at the places of contact among solids). The overall stability of the dispersed system may be influenced by the interplay of several phenomena occurring simultaneously, e.g. adhesion, chemihesion, capillary pressure, etc.629,630,634 Amorphisation is often accompanied by a condensation process within the crystal lattice. This is typical for kaolinite and it is shown in Fig. 36.629
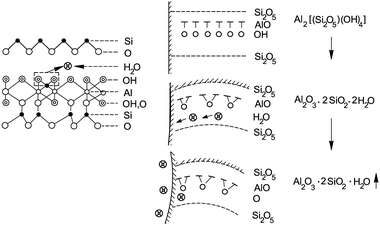 | ||
| Fig. 36 Amorphisation by internal condensation. Left: water loss from the lattice of kaolinite; right: scheme of amorphisation by lattice distortion and water loss. Reprinted with permission from ref. 629. Copyright 1998, Taylor & Francis. | ||
Interactions between solid particles performed under mechanical stress and simultaneous capillary condensation are denoted as mechanochemical capillary reactions.643 Alteration in wetting properties,644–646 modification of surface charge and zeta potential,647,648 dissolution/leaching of surface/amorphous phase,630,647 change in ion-exchange and sorption properties,629,630,649 and rheological character635,638,650,651 are all reported due to the presence of water during milling.
The presence of water as a reaction product or through extraneous addition changes the regime of interaction between the solid phases involved in mechanochemical reactions. The flow and transport properties of the system change as a whole and dissolution/precipitation reactions can be promoted. Numerous examples of mechanochemical reactions are available where different roles of water are elucidated (e.g.ref. 629–634). Selected examples are presented here. Hydrothermal aspects of mechanical activation and wet milling in a stirred ball mill (attritor) which involves extraneous water addition are presented subsequently.
Mechanical activation of copper sulphate (CuSO4·5H2O) and calcium sulphate hemihydrate (CaSO4·0.5H2O) has been reported.630 During vibration milling, a water condensation reaction occurs. When open milling is utilized, water molecules are able to escape. When closed milling is applied, the dissolution and recrystallisation occurs, i.e. reformation of CuSO4·5H2O crystals. Mechanical activation of the calcium sulphate hemihydrate follows the sequence: CaSO4·0.5H2O → CaSO4·2H2O → CaSO4. Condensation water dissolves calcium sulphate which precipitates as the dihydrate and eventually decomposes into anhydrite and water. Mechanical activation of hydroxides and oxyhydroxides – Ca(OH)2, Mg(OH)3, Al(OH)3, γ-Al(OOH)629–631,652–658 – and silicates – kaolinite, monmorillonite, illite, talc, mica, etc.629–631,659–667 – which undergo mechanically induced dehydroxylation [2(ROH) → R2O + H2Ovapour] has been extensively studied. The dehydroxylation reaction requires proton transfer which is possible if one of the OH− ions is broken.652 The rate of dehydration and amorphisaton follows the sequence: Al(OH)3 > Ca(OH)2 > Mg(OH)2, even though all three hydroxides have the same structure comprising closely packed OH− sheets. Higher amorphisation of Al(OH)3 is explained in terms of structurally conditioned vacancies which are absent in other hydroxides.652 The kaolinite (Al2(Si2O5)(OH)4) surface hydroxyl groups lost during milling were replaced with coordinated and adsorbed water on the surface of the octahedral sheet.660 Hydrogen release during dry milling of kaolinite was reported by Kameda et al.663 H2 is formed by the reaction between surface water molecules and mechanoradicals created by the rupture of Si–O or Al–O–Si bonds.663,664 Carbonation of Ca(OH)2 is facilitated by water which may be present as humidity or as a consequence of water condensation during mechanical activation. The carbonation of CaO and MgO during milling required a continuous flow of water.629 Sydorchuk et al.637 studied the mechanochemical activation of fumed silica (non-porous), aerosilogel and silicagel (porous silicas) in air and in the presence of water and alcohol (solid : liquid ratio 0.2) using planetary milling. An extensive decrease of specific surface area and destruction of the pore structure of aerosilogel and silicagel was observed during dry milling. On the contrary, milling in water caused minimum changes in specific surface area and total pore volume. The liquid phase milling of non-porous aerosil leads to formation of a porous substance similar to aerosilogel which possesses mesoporous (after milling in water) and macro-mesoporous structure (after milling in alcohol). The method of boehmite preparation (γ-AlOOH) is found to influence its mechanical activation.658,667 Boehmite (γ-AlOOH) prepared by thermal decomposition of gibbsite showed an anomalous decrease in its surface area from 264 m2 g−1 to 65 m2 g−1 after 240 min of planetary milling.658 In sharp contrast, boehmite prepared by hydrothermal transformation of gibbsite showed an increase in specific surface area from 3 m2 g−1 to 35 m2 g−1 under identical conditions of milling.668 The mechanically induced dehydroxylation in high-surface-area boehmite is accompanied by coalescence of fine pores (2 and 4 nm) to form bigger pores (20 nm) and an aggregation process. Dehydroxylation of a low-surface-area boehmite sample occurs with a simultaneous increase in density of pores having size centered around 4 and 20 nm and the formation of loosely bound aggregates.
Juhász and Opoczky have highlighted the importance of liquid bridges629,634 which provide an ideal environment for dissolution, species transport and precipitation reactions. Dissolution and precipitation reactions were hypothesized during mechanical activation of gypsum–aluminum oxide and magnesium oxide–kaolinite mixtures. Solid bridges formed due to precipitation reactions are continuously broken and replaced by new virgin liquid bridges. In some cases (e.g. CuSO4·5H2O–BaCO3 system), freezing of the reaction was reported due to large volume of precipitate which occupies the entire volume with bridges.629
The hydrothermal aspect of mechanochemical treatment invoked significant interest among Japanese and Russian researchers around the 1970s. Kiriyama et al.669 noted, as early as in 1968, the similarity between spinel ferrites formed under mechanochemical conditions and hydrothermal conditions. Assuming reactant particles form a porous matrix containing liquid, Boldyrev670 made theoretical estimates of the temperature rise from viscous flow in the pores, adiabatic compression of the fluid, elastic and plastic deformation of solid phase and friction at the contact between solids. Adiabatic compression of the gas bubbles in the fluid (due to impulses of pressure) and the friction at the contact points may give rise to significant increase in temperature and pressure. The localized temperature may increase up to 750 °C and the maximum pressure could reach as high as 450 atm or more.633,670,671 Consequently, the thermodynamics of the local reaction environment favors reactions which may otherwise be kinetically inhibited at the bulk system temperature and pressure due to rate-determining steps such as interfacial reaction, crystal dissolution, dehydroxylation, diffusion, crystallization, etc.672–675 Also, micro-autoclaves can be formed due to (a) device- and pulp-specific circumstances, (b) collision of two solid samples with rough surfaces and (c) compression of liquid in capillary pores (Fig. 37).633 Recent reviews and updates are presented by Temuujin,674 Boldyrev633 and Billik and Čaplovičová.632
 | ||
| Fig. 37 Some micro-autoclave formation possibilities: (a) device- and pulp-specific circumstances, (b) collision of two solid samples with rough surface and (c) compression of liquid in capillary pores. Reprinted with permission from ref. 633. Copyright 2002, Elsevier. | ||
A large number of examples which demonstrate the possibility of synthesizing hydrothermal phases by a mechanochemical route can be cited, for example afwillite (Ca3(SiO3OH)2·2H2O), tobermorite (Ca5(OH)2Si6O16·4H2O),633,676,677 hydenbergite (Ca,Fe)Si2O6, calcium hydrosilicate phases,633 products resembling talc or chrysotile,674,678 PbTiO3,672 hydroxyapatite,679–682 hydrated rubidium thio-hydroxosilicogermanates [RbzGexSi1−xSz(OH)4−z·yH2O (2 ≤ z ≤ 3; 0 < y < 1.7)],683etc.
Unlike solid–solid mechanochemical reactions similar to hydrothermal synthesis, the composition of the phases formed under mechano-hydrothermal conditions is also determined by the mole fraction of the components taken for synthesis.631,633 There is no clear generalized view if the formation of hydrothermal phases can a priori be predicted. The role of the quantity and form of water has been highlighted in the literature.633,678,684 Based on the ratio (J) of total volume of water present in pores (W) and total pore volume (Wmax), i.e. J = W/Wmax, Boldyrev633 indicated that hydrothermal conditions can prevail in the regime 0.85 < J < 1 (hydrodynamic continuity of the liquid exists, but it is disturbed under the mechanical action applied on the pulp). If J < 0.85 (the liquid is not hydraulically continuous and the mechanical pulse acts directly on the solid phase) and if J > 1 (the liquid is hydraulically continuous), no local increase in temperature occurs. Examples of the synthesis presented by Boldyrev633 can be divided into 2 groups: (i) hydrothermal processes performed with 1–5% water added from outside (e.g. synthesis of hydenbergite (Ca,Fe)Si2O6, and tobermorite Ca5(OH)2Si6O16·4H2O) and (ii) the reactions in which water appears in the system as a result of dehydration of one of the components in the mixture (e.g. preparation of calcium hydrosilicates from the mixtures containing Ca(OH)2 and SiO2·H2O). The importance of the optimal water content for the hydrothermal reaction mechanism was verified for Mg(OH)2, SiO2·nH2O, Mg(OH)2 and silicagel mixtures.678b Kosova et al.671 found that the molar ratio of CaO–SiO2 in mixtures of Ca(OH)2 and SiO2·0.5H2O is an important factor in the hydrothermal reaction mechanism. The chemical form of reactants (e.g. M(OH)2–SiO2, M(OH)2–SiO2·nH2O or MO–SiO2·nH2O, where M = Ca, Mg) plays an important role in the synthesis of hydrothermal phases by a mechanochemical route.633,674
A number of studies have focused on mechano-hydrothermal synthesis of hydroxyapatite (HAp).679–682 Suchanek et al.681 compared the nature of HAp prepared by hydrothermal and mechano-hydrothermal synthesis. Hydrothermal synthesis yielded well-crystallized needle-like HAp powder (size range 20–300 nm) with minimal levels of aggregation. In sharp contrast, room-temperature mechanochemical–hydrothermal synthesis resulted into agglomerated, nano-sized (20 nm), mostly equiaxed particles regardless of whether the HAp was stoichiometric, carbonate-substituted (CO3HAp) or contained both sodium ions and carbonate (NaCO3HAp). The slurry comprising of Ca(OH)2 and CaCO3 (or Na2CO3) and (NH4)2HPO4 contained 13 wt% solid. It means it falls in the regime (J > 1) where temperature and pressure rise is not expected. The mechanochemical-hydrothermal synthesis of HAp was carried out in a multi-ring media mill at room temperature. These results were not reproduced during wet milling of dicalcium phosphate dihydrate and calcium oxide (5–100 ml water/15 g mixture) in a planetary mill,684 highlighting the importance of constituents, mill type and water content. Recent studies685 have incorporated the use of emulsions in mechanochemical-hydrothermal reactors for better regulation of nucleation and growth, which resulted in HAp that was far less aggregated.
One of the important features of mechanochemical activation in the presence of water is the enhanced solubility.631,633,678,686 The mechanically activated solids can also show unusual response during post-activation reaction. During hydration of mechanically activated blast furnace slag in an attrition mill, it has been found that the crystallinity of calcium–silicate–hydrate phases (CSH, C = CaO, S = SiO2 and H = H2O) increases with an increase of milling time. The hydration product also showed the presence of α-C2SH phase, which is known to form only under hydrothermal conditions.647
Our understanding of hydrothermal reactions under mechanochemical conditions is still very limited. The use of a mechanochemical-hydrothermal approach has the potential to take hydrothermal technology to new and totally unexplored heights.
The amount of added extraneous water may vary from a few percents to a few tens of percents. The intensity of mechanochemical processes involving the formation of liquid bridges can be improved by pumping liquid vapours through the mill or material.629 Liquid-assisted grinding LAG (or solvent-drop grinding) is used in organic mechanochemistry and the significance of the amount of water is highlighted in the literature.687 For milling in kneader and pug mills, solid–water paste is used for mechanical activation and the amount of solid may be 70% and sometimes even more.635 Milling in wet conditions is generally considered more efficient in terms of energy consumption in comparison with dry milling.630 Iwasaki et al.688a,b used mechanical activation of freshly prepared suspension of co-precipitated mixture of Fe(OH)2 and α-FeOOH in argon atmosphere using a ball mill to synthesize nanosized super-paramagnetic magnetite particles. The reaction kinetics was favoured by milling energy (rotation speed).688a In a subsequent paper, the same group of researchers689 reported direct synthesis of nanosized magnetite starting from α-FeOOH. The hydrogen formed due to the rusting of media/mill reduces the ferric iron to ferrous state which is then hydrolyzed to Fe(OH)2 and participates in the reaction of magnetite formation. Even though, in specific instances, traditional ball milling can be used for mechanochemical synthesis, the scope is limited due to low milling energy. The presence of water and high milling energy in an attrition mill provide unique conditions for fine milling and mechanical activation.647,648,690
In conclusion, we will focus specially on wet milling in an attritor due to its technological significance. The increase in size of these mills from 500 L in 1992 to more than 10000 L in recent times, imparts special significance to these mills for large-scale operations.634,691 Baláž has reviewed the historical development and applications of attritors.6 Attrition mills or stirred ball mills differ from conventional ball mills in terms of media diameter (it is much smaller, generally 2–4 mm, but it may be as small as 50 μm in the case of nanomilling634,692), which imparts greater media–material contact, and a rotating impeller (speed up to 4500 rev min−1) which imparts high kinetic energy to the media. The milling energy can be controlled through a judicious selection of media (size, density) and rotation speed.648,693 Fine milling and mechanical activation effects in attrition milling are exploited in developing a number of hydrometallurgical processes and novel building materials.6,634,691,694,695 Currently, several hundred installations are in use in commercial mineral processing operations.691,696 In general, attrition milling is used for fine milling. The milling in an aqueous environment and/or the use of small balls is more favourable for new surface formation, whereas dry milling and/or use of larger balls favours amorphisation.634 Attrition milling has been successfully used to produce nano-sized materials, e.g. ferrofluids,697 through media selection and dispersion control.692,698–700 The utilization of an attrition mill, which is generally considered as fine-milling device, can result in interesting mechanochemical effects as observed for a large number of minerals and materials, for example basic metal sulphides (e.g. chalcopyrite, tetrahedrite, enargite, pentlandite, etc.),634,694 Al-oxyhydroxide minerals (gibbsite, boehmite),701–703 alumina,638,651,699,704–707 calcium carbonate,708 olivine,636 kaolinite,635 wollastonite,645 coal,709 pigments,700 bauxite,710–715a cement715b and waste materials (slag, red mud).647,716 Fritsch704 and Oberacker et al.707 observed that while undergoing wet milling a change of phase composition from α-alumina to an aluminum tri-hydroxide phase can occur. Kikuchi et al.706 examined the change of lattice deformation and lattice strain in α-alumina in a wet-milling process over a period of time. Stenger et al.699 observed the transformation of α-Al2O3 to bayerite Al(OH)3. The formation of bayerite was found to depend on temperature and the opposite phase transformation was also observed during the process. This was accompanied by formation of meso- and possibly micro-pores. Alex703 made similar observations for the transformation of boehmite (γ-AlOOH) to bayerite. In a few studies, milling in an attrition mill is compared with milling in other milling devices. Particle morphology of milled gibbsite in attrition and jet mills revealed that under attrition the main mechanisms were the rupture of grain joints leading to the dissociation of crystallites and the chipping and breakage of the crystallites. Under impact stress (as in a jet mill), the fragmentation mechanisms included concomitant rupture and breakage, which produce a more uniform-sized product.701 Baudet et al.635 observed that delamination and transverse breakage occur sequentially during stirred media milling and simultaneously in high-energy kneading in a pug mill. Baláž et al.636 compared the mechanical activation of olivine in attritor and planetary mills. It can be inferred from the presented data that for the same amount of energy input (2 kW h kg−1), the surface area and percentage of crystalline fraction remaining was 34 m2 g−1 and 13% for attrition milling and 4.7 m2 g−1 and 37.4% for planetary milling.
Milling energy and the presence of water are critical parameters to achieve mechanical activation during attrition milling. It has been reported that attrition-milled blast furnace slag is completely hydrated after 28 days.647 This is in sharp contrast with the slag which is continuously milled in a conventional ball mill for nearly the same length of time and showed only 15–20% hydration.690,717 Isothermal conduction calorimetric studies revealed distinctly different hydration behaviour for the attrition-milled (high energy, wet) and vibration-milled (high energy, dry) slag of same size (12 μm) – attrition-milled slag was hydrated, whereas vibration-milled slag did not show signs of hydration.647,690 The effect of milling energy on the amorphisation of gibbsite during attrition milling has been reported.648 The presence of a hard phase during milling of a softer phase, e.g. hematite and quartz in gibbsite milling, can accelerate and enhance the amorphisation of the soft phase.
Surface chemistry plays an important role during attrition milling. Ding et al.708 observed that surface modification of calcium carbonate from hydrophilic to hydrophobic was improved if the modification by sodium dodecylsulphate (SDS) is carried out in an attrition mill. The surface of wollastonite was also changed from hydrophilic to hydrophobic during attrition milling in the presence of titanate.645 Alex et al.648 reported that during attrition milling of gibbsite, surface charge and zeta potential is altered due to loss of texture (since charge on the faces and edges of gibbsite crystals differs) and amorphisation.648 Zeta potential change has also been reported during attrition milling of granulated blast furnace slag (amorphous alumino-silicate).647 This change is attributed to enhanced leaching from the surface during milling. In the course of attrition milling in the micrometer to nanometer range, particle–particle interactions become increasingly important.651,692,698,718,719 The suspension stability determines the equilibrium state of milling, agglomeration and deagglomeration.698 The particle–particle interaction and stability can be controlled through the change in surface charge by pH control and/or by addition of dispersant so that the repulsive force dominates over weak van der Waals forces.719
7. Organic–inorganic interphase
7.1 Organic crystals with their interface to organic and inorganic species
Mechanochemical phenomena of organic crystals. Organic crystals (OCs) comprise molecules among which non-covalent interaction predominates. They tend to deform inelastically, often leaving deformed molecular states quenched. Mechanically forced molecular deformation induces a decrease in the HOMO–LUMO gap and tends to trigger a chemical reaction.720 The concept is understood under the general concept of inverse Jahn–Teller effects.720,721 Unlike the traditional stream of mechanochemical studies based on mineralogy and associated solid-state chemistry,5e,6,722 organic mechanochemistry has mainly been driven by interest in organic synthesis aimed at particular bond cleavage.723 In the meantime, comprehensive review articles have appeared centered on organic compounds with extended wider views. Beyer et al.8f tried to put mechanochemical phenomena into perspective with modern tools like atomic force microscopy and quantum molecular dynamics simulation. Boldyreva724 also tried to generalize the relationship between mechanical action and chemical interactions, associated with the mechanical properties of individual bonds in single molecules. James et al.725a emphasised the importance of co-crystals in their comprehensive review. The crystals were defined as multi-component molecular crystals. Formation of metal–ligand bonds is one of the typical mechanochemical reactions with OCs.725b–d A series of experimental works in the field of organic synthesis under the concept of mechanochemistry were carried out, with the primary interest of solvent-free fast reactions, e.g. for domino oxa-Michael-aldol reaction725e or Morita–Baylis–Hillman reaction.725f Diels–Alder reactions in a solid state between anthracene (AN) derivatives and p-benzoquinone (BQ) under mechanical stressing were accelerated by adding a catalytic amount of 2-naphthol (NP) or (rac)-1,10-bis-2-naphthol (BN).726,727 Formation of the charge-transfer complex with strong hydrogen bonds was a key issue, since BN is capable of incorporating BQ together with AN derivative.
Change in the properties of organic compounds in the presence of inorganic crystals. Mechanical stressing on organic compounds in the presence of metal oxides (MOs) results in various unusual interactions and alters the properties of both parties.728 An example is given below in the case of immobilization of drugs by milling with silica nanoparticles.729 As indomethacin (IM) was co-ground with silica nanoparticles, bridging bonds were formed. Interaction of OH in IM and O of silica and consequent formation of C–O–Si bridging bonds were confirmed by Si2p XPS (Fig. 38) and simple molecular orbital simulations.
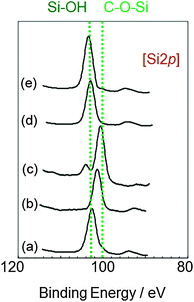 | ||
| Fig. 38 Si2p XPS spectrum of indometacine (IM) and silica (S), with milling time in min. (a) IM-S0, (b) IM-S30, (c) IM-S180, (d) S30 and (e) S180. Reprinted with permission from ref. 729. Copyright 2004, LAVOISIER SAS. | ||
Anion exchange of metal oxides with the aid of organic crystals. Co-milling of OCs and MOs enables anion exchange in the MOs, i.e. oxygen with nitrogen, carbon, sulphur or halogens, altering electronic states of MOs. Since this inevitably changes the absorption spectra of MOs, introduction of various functionalities is expected with respect to the photonic properties. Associated change in the defect structures of MOs plays an important role in subsequent solid-state processes. Several case studies will be explained below.
Substitution of oxygen in TiO2 with fluorine of poly(tetrafluoro ethylene), PTFE, took place by co-milling of the dry mixture comprising TiO2 with 10 wt% PTFE.730 The entire process is triggered by the partial oxidative decomposition of PTFE, accompanied by the abstraction of oxygen from the TiO2. This, in turn, results in the formation of the diverse states of locally distorted coordination units of titania, i.e. TiO6−nVon, located in the near-surface region, where Vo denotes oxygen vacancy. Introduction of F is understood as a subsequent partial ligand exchange between F and O. The reaction scheme is shown in Fig. 39.
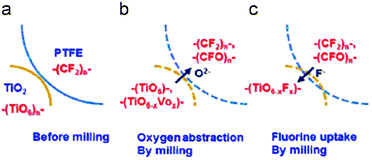 | ||
| Fig. 39 Schematic representation of mechanochemical anion exchange of O in TiO2 with F by co-milling with PTFE. Reprinted with permission from ref. 730. Copyright 2012, Elsevier. | ||
Faster mechanochemical anion exchange of oxygen with fluorine was observed as SnO2 was co-milled with poly(vinylidene fluoride) PVdF. The changes in various spectra (FT-IR, Raman and 19F-MAS NMR) occurring during co-milling consistently indicated that PVdF has been decomposed at the early stages of co-milling with SnO2. The incorporation of fluorine from PVdF into SnO2 was unambiguously exhibited by the changes of XPS F1s and Sn3d peaks, as shown in Fig. 40, where the intensity of the binding energy peak specific to F in PVdF at around 887.8 eV decreases while that close to SnO2-F at around 685.3 eV increases. The reaction mechanisms triggered by the mechanochemical decomposition of PVdF to incorporate F into SnO2 exhibit close proximity to those observed on the system TiO2-PTFE mentioned above.
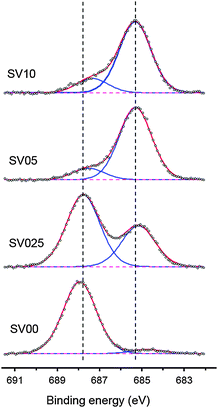 | ||
| Fig. 40 F1s XPS of the co-milled mixture of SnO2 and PVdF from ref. 16. SV00, 025, 05 and 10 denote milling time 0, 15, 30 and 60 min, respectively. | ||
7.2 Application to ceramics processing
Wet mechanochemical process is applicable to introduce N into TiO2, where a mechanochemical reaction takes place at the boundary between an aqueous solution of glycine and TiO2 nanoparticles. The principal mechanism is the formation and deposition of complexes, from which nitrogen can diffuse into TiO2 upon subsequent annealing. It is worth noting that the product exhibited promising antibacterial photocatalytic activity upon irradiation of blue light, peaking at around 440 nm, as exhibited in Fig. 41.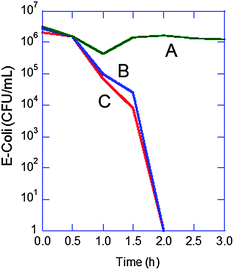 | ||
| Fig. 41 Change in the bacteria (E. coli) concentration with the blue light emission time. A: pristine TiO2, B and C: co-milled with glycine solution and annealed at 500 °C. Reprinted with permission from ref. 156. Copyright 2012, Cambridge University Press. | ||
Increase in the rate of solid-state reaction is another important aspect of mechanochemistry with respect to the interaction between OCs and MOs. Two examples are given below. Barium titanate (BT) remains one of the most important key materials in the field of electronic ceramics. Co-milling of the starting materials of BT solid-state synthesis with a series of nitrogen-containing organic compounds brings about better reactivity and accordingly high functionality, represented by, for example, tetragonality of BT. When the starting materials of BT, an equimolar mixture of TiO2 and BaCO3, were vibro-milled by nylon-coated steel balls in a PTFE vial, finer BT particles were obtained due to the homogenized reaction mixture.731 One obvious reason was the particle size reduction of the reactants without causing severe agglomeration, as shown in Fig. 42, in spite of dry processing. This can partly be attributed to the abrasion of nylon, serving as surface modifier to prevent agglomeration.
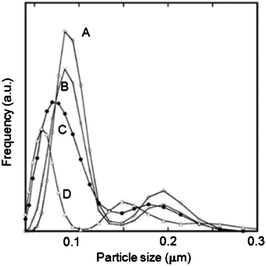 | ||
| Fig. 42 Change in the particle size distribution of the reactant for BT with milling time: A: 0; B: 2 h; C: 5 h; D: 10 h. Reprinted with permission from ref. 732. Copyright 2004, EDP Sciences. | ||
Further suspecting the chemical role of nylon, other nitrogen-containing organic compounds were added. As shown in Fig. 43, addition of glycine (Gly), one of the typical amino acids, was effective to obtain phase-pure perovskite after short co-milling.732
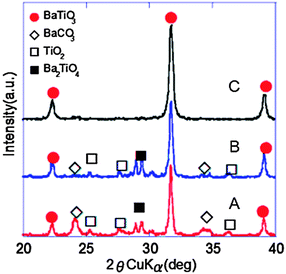 | ||
| Fig. 43 X-ray diffractograms of the equimolar mixture of TiO2 and BaCO3 after calcining in air at 850 °C for 2 h. A: intact mixture; B: with 1 wt% Gly; C: with 1 wt% Gly and subsequently vibro-milled for 0.5 h. Reprinted with permission from ref. 732. Copyright 2004, EDP Sciences. | ||
Due to the elevated reactivity by adding a small amount of Gly and co-milling, higher tetragonality of BT was attained, compared at the same particle size (Fig. 44). The influence of Gly on the soft mechanochemical treatment was further examined by wet ball milling of the reacting mixture comprising TiO2 and BaCO3 with Gly up to 9 wt%.734 By using high-temperature XPS, it was concluded that Gly residue persists up to around 350 °C and destabilizes Ti–O bonds in TiO2 prior to the initiation of reaction toward BT. This, in turn, leads to a decrease in the reaction termination temperature due to higher mobility of Ba2+ ions through the reaction zone.
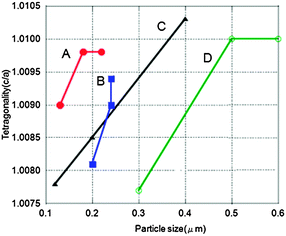 | ||
| Fig. 44 Relationship between tetragonality and particle size of BaTiO3, obtained after heating at 1050 °C, from the starting mixtures. A: intact, B: dry vibro-milled for 10 h, C: with 0.5 wt% Gly and wet vibro-milled for 30 min.732 Curve D was cited for comparison, from ref. 733. | ||
A similar principle works on more complicated reaction systems, e.g. for the phase-pure solid-state synthesis of Li4Ti5O12 (LT45), a promising anode material for Li-ion batteries due to its zero-dilation upon charge and discharge cycles.735 In this case, mechanical activation at the beginning did not work well, because of the extraordinary stability of the intermediate Li2TiO3 (LT21). Therefore, the intermediate mixture, i.e. LT21 with a slight excess of TiO2 based on the LT stoichiometry, was mechanically activated in the presence of an amino acid. Out of the 3 species of amino acids, alanine gave the best result with phase purity of 98% and average particle size of 70 ± 20 nm.
7.3 Application to biological imaging
Research on fluorescent semiconductor nanocrystals (also known as quantum dots) has evolved from electronic materials science to biological applications. In 2005, a review paper was published dealing with the synthesis, solubilization, functionalization and applications of quantum dots to cell and animal biology.736 Mechanochemistry represents an interesting method for the preparation of nanocrystalline materials.737–739Cadmium selenide (CdSe) exerts unique fluorescent properties and with its quantum confinement it is perfectly suitable for applications in biological imaging.740 However, CdSe particles contain toxic cadmium which represents a health hazard.741 One of the ways of preventing the toxic effect of cadmium is to provide a protective shell around the CdSe nanoparticles. ZnS is fairly suitable for this application, because of its several properties including a wider bandgap than CdSe.742 In addition to lower cytotoxicity, the addition of ZnS leads to the increase of the stability of material, the improvement of optical properties of the system and the decrease of the surface oxidation.743,744
In addition, CdSe/ZnS nanoparticles are bioconjugable.745–747 The utilization of amino acids containing sulphur as bioconjugation agents are of particular interest. The metal complexes of sulphur-containing amino acids are interesting because of their unusual electron-mediating abilities and potential biological functions.748
An interesting work was done dealing with the capping of these CdSe/ZnS nanoparticles with L-cysteine, whose water solution was used.749 Not only have the authors obtained a nanocomposite structure instead of the typical core–shell structure, but also the transformation of capping agent (cysteine to cystine) by the effect of mechanical action occurred.
The proof of cystine presence in the nanocomposite was given by FTIR (Fig. 45). In the spectrum of pure L-cysteine (Fig. 45a), the most important peak around 2550 cm−1 corresponds to the stretching vibration of the –SH group. Also the peaks for –COO− and –NH3+ groups are present. In the spectrum of pure L-cystine (Fig. 45b), peaks for –NH3+ and –COO− group are also present. Of course, the peak for –SH group is missing.
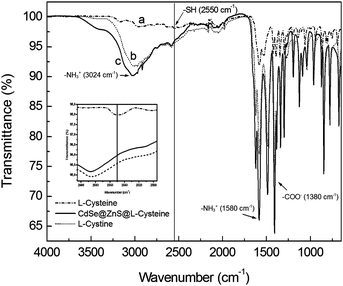 | ||
| Fig. 45 FTIR spectra of cystine-bioconjugated CdSe@ZnS nanocomposites. Reprinted with permission from ref. 749a. Copyright 2012, Acta Physica Polonica A. | ||
After the comparison of the spectra of synthesized CdSe@ZnS co-milled with L-cysteine (Fig. 45b) and of pure L-cystine (Fig. 45c), it is clear that they are the same. The authors concluded that L-cysteine is oxidised to L-cystine during the milling process and that this transformation occurs because of the presence of air and water during milling. However, the exact type of bond present between the capping agent and the particles, or in the particles themselves (bonds between CdSe and ZnS), is not clear and has to be elucidated in future mechanochemical studies.
8. Industrial aspects
This chapter is devoted to the application of mechanochemistry in materials engineering, heterogeneous catalysis and extractive metallurgy. Further applications in minerals processing, chemical engineering, the building industry, the coal industry, agriculture, pharmacy and waste treatment can be found in ref. 7. In some of these processes, nanoparticles with extraordinary properties are formed and this predisposes the obtained materials to non-traditional applications. The environmental aspects of these processes are particularly attractive.722,750,751The main advantages in comparison with the traditional technological procedures are:
• decrease in the number of technological stages,
• exclusion of operations that involve the use of solvents and gases, and
• the possibility of obtaining a product in the metastable state which is difficult (or impossible) to obtain using traditional technological procedures.752,753
Simplification of the processes, ecological safety and extraordinariness of the product characterize the mechanochemical approach in technology.
8.1 Materials engineering
Non-equilibrium processing of materials has attracted the attention of a number of scientists and engineers due to the possibility of producing better and improved materials than with conventional methods.755 The technique of mechanical alloying (MA) was used for industrial applications from the beginning and the basic understanding and mechanism of the process is beginning to be understood only now. One of the greatest advances of MA is in the synthesis of novel alloys, e.g. alloying of normally immiscible elements, which is not possible by any other technique. The MA products find applications in various industries and these are summarized in Fig. 46. Mechanical alloying for commercial production is carried out in mills of up to more than 1000 kg capacity. Ball mills to produce oxide-dispersion-strengthened nickel- and iron-based alloys are used at facilities in the USA and Great Britain (Fig. 47). | ||
| Fig. 46 Typical current and potential applications of MA products. Reprinted with permission from ref. 754. Copyright 2001, Elsevier.754 | ||
 | ||
| Fig. 47 Commercial production-size ball mills used for mechanical alloys. Reprinted with permission from ref. 14. Copyright 2001, Elsevier. | ||
The development of new production technologies for NiTi shape memory alloys is always challenging. Recently, two powder metallurgical processing routes involving mechanical activation of elemental powder mixtures and densification through extrusion or forging were introduced.756 Those processes were named mechanically activated reactive extrusion synthesis (MARES) and mechanically activated reactive forging synthesis (MARFOS). Heat treatment was performed in order to adjust the B2-NiTi matrix composition, yielding a microstructure consisting of a homogeneous dispersion of Ni4Ti3 precipitates embedded in nanocrystalline B2-NiTi matrix.
The MARES process, as an innovation in powder sintering, was used for the first time to produce NiTi.757–759 Equimolar powder mixtures of elemental Ni and Ti were firstly mechanically activated for 4 hours and then extruded at relatively low temperatures (between 300 and 600 °C). No intermetallic phase formation was observed after mechanical activation. On the contrary, crystalline phases (Ni and Ti) and an amorphous phase were observed. The end product was constituted of agglomerated microsized particles containing inhomogeneous areas of Ti and Ni. Differential thermal analysis of the as-milled powders showed two small exothermic peaks at about 460 and 540 °C, corresponding to the structure relaxation of the amorphous phase and to the synthesis of Ni–Ti intermetallics, respectively. The outcome of MARES trials was sensitive to the extrusion temperature, ram speed and to the total heating time prior to the application of load. The microstructure of the extruded material consisted of NiTi, NiTi2 and Ni3Ti besides Ni and Ti. MARES results are very encouraging for the formation of Ni–Ti intermetallics through a controlled synthesis reaction.
The production of alloys through MARFOS process from elemental powder mixtures with equiatomic composition has been investigated for the first time by Neves et al.760 The new powder metallurgy processing was successfully applied to the production of bulk NiTi alloys and can be considered as a promising technique for producing other bulk intermetallic compounds. With MARFOS, almost fully dense (99.4%) products with multiphase nanocrystalline structure have been produced. This was possible because mechanical activation, besides modifying the reactivity of the pristine powders, also produced powder mixtures with hot deformation ability, allowing the densification to be achieved by forging at relatively low temperature (700 °C). The short duration of mechanical activation (4 hours) successfully produced powder mixtures capable of being hot-deformed at relatively low temperatures. As a consequence, bulk product composed of intermetallic and metallic nanocrystalline phases was effectively obtained at 700 °C. An additional heat treatment at 950 °C for 24 hours removed almost all the undesired phases and maintained the nanocrystalline structure, resulting in an Ni-rich NiTi matrix. The formation of such a microstructure may be attributed to the presence of oxygen and nitrogen impurities in the materials. After ageing at 500 °C for 48 hours, some degree of compositional rectification was achieved in the NiTi matrix due to the formation of Ni4Ti3 precipitates.
The preparation of highly dense bulk materials with a grain size in the range from a few to hundreds of nanometers is currently the objective of numerous studies.761 This has been achieved in this regard by using the methods of mechanically activated field-activated pressure-assisted synthesis (MAFAPAS), which has been patented,762 and mechanically activated spark plasma sintering (MASPS). Both methods, which consist of the combination of mechanical activation step followed by consolidation step under the simultaneous influence of electric field and mechanical pressure, have led to the formation of dense nanostructured ceramics, intermetallics and composites, such as MoSi2,763,764 FeAl,765–772 and NbAl3.773,774
8.2 Heterogeneous catalysis
Heterogeneous catalysis is the phenomenon of the enhancement of the rates of chemical conversions in fluids by the presence of solid surfaces, which are called catalysts.775 Catalysts are tools that have a great impact on industry and environmental protection. They are used in the manufacture of a huge amount of goods and materials each year, including energy, chemicals, pharmaceuticals, and plastics.776,777 Catalysts play a key role in everyday life improvement, environmental protection, automobiles and industrial exhaust gas cleaning, purification of soils and ground waters. They have been generally developed by using inefficient “trial and error” methods. Catalysts have been intensively investigated, and the main goal is to create general theories that would make it possible to predict the catalytic properties of solids.4f,14,194,777–781Nano-catalysts seem to be the next generation catalysts as they set a new paradigm in catalysis. Nano-catalysts are very promising, as they possess some of the main characteristics of the “perfect” catalyst, such as high dispersity (including high specific surface area) and a great core/shell ratio (which gives rise to the presence of highly active unsaturated chemical bonds). In addition, they can be designed and engineered at the atomic scale by controlling their structure. This leads to the improvement of their catalytic properties (efficiency, lower amount of by-products and waste), prolongation of their life (deactivation and corrosion stability) and reduced catalyst cost.14,194,777–781 Catalysts are specific products and a number of requirements should be taken into account during their synthesis. Contact area, mass transfer processes and activation barriers limit the rate of chemical interactions. In this regard, conventional methods used for synthesis of heterogeneous catalysts have a number of significant disadvantages, such as performing processes at high temperatures and pressures or in solution, etc.
Some of the main properties of mechanochemistry are very suitable for highly effective catalyst synthesis either as a step or as the main stage of preparation. Mechanochemistry is one of the most interesting techniques from an industrial point of view; it is one of the least sophisticated and inexpensive technologies, especially in the case of nanosized catalyst preparation.4f,14,194,778,780,781 Mechanical activation changes the overall reactivity of solids and allows running of catalytic reactions through new pathways at nearly ambient conditions.780,781 The main concepts of the effect of mechanochemical activation (MCA) on the activity and selectivity of catalysts have been theoretically stated, but a number of questions remain unclear and controversial.14,194,778,780 Mostly, they reflect the dependence of catalytic action of a solid on the parameters of mechanochemical treatment (type, intensity, duration, medium, etc.) in a studied chemical reaction. MCA changes the chemical nature and structure of a solid as well as the catalytically active sites of the main and side reactions. Correct activation conditions are the most important factors positively influencing the selectivity towards the desired product. Stability, resistance, lifetime and deactivation of catalytically active sites are also significantly dependent on the treatment conditions. A solid catalyst accumulates excess potential energy during MCA. Elastic and plastic deformations as well as a great variety of defects are present. This leads to an increase in its reactivity. Its thermodynamic potential tends to decrease, which can be a consequence of different relaxation channels. A number of important phenomena occur in the bulk and on the solid surface, but the latter becomes critical in the case of catalytic action. A non-equilibrium system is responsible for the formation of a large variety of structural, energetic, chemical and physical properties depending on the MCA intensity. Under MCA conditions, the energy of the reacting phases significantly differs from reference data corresponding to standard conditions. For this reason, non-equilibrium systems can appear under such conditions. As the energy stops dissipating, such energy-intensive systems undergo an extinction relaxation process towards standard characteristics. Relaxation decays and complete equilibrium is not attained and a highly active metastable non-equilibrium catalytic system appears.14,194,778–781
Mechanochemical synthesis (MCS) is an alternative preparation method, which boosts the main concepts of a green chemistry approach. Green chemistry involves innovation in chemical research and engineering that encourages the design of processes to minimize the use and production of hazardous materials and also to reduce the use of energy. These requirements are fulfilled by preventing or minimizing the use of volatile and toxic solvents and reagents, minimizing chemical wastage, development of atom-economical processes and recyclable supported catalysts that are less toxic, biodegradable and can be used at low loading. The basic approaches to achieve the objectives of green chemistry include the design and synthesis of non-traditional catalysts and fundamentally new chemical processes utilizing them, as well as the use of non-traditional methods for activation of chemical processes (microwave radiation or mechanochemical activation of reagents/catalysts).782
The development of MCS of catalysts concerns the preparation of solids with desired properties such as phase composition, crystallization degree, specific surface area, porous structure, defectiveness, dispersion, morphology, thermal stability, structural and mechanical properties, component distributions in the case of supported samples, etc. MCA is efficient in creating waste-free energy-saving methods for the preparation of hydride catalysts, heteropoly acid catalysts, and catalysts for hydrocarbon decomposition into hydrogen and carbon materials, as well as for the synthesis of previously unknown catalytic systems.780a Significant results can be achieved by applying MCA in industrial catalyst manufacture for:779
• preparation of finely dispersed and nanosized systems,
• partial or complete substitution of synthetic stages in which solvents are used (for prevention of ecologically harmful wastes),
• synthesis of new and non-equilibrium solid catalysts that cannot be prepared by traditional methods (for example, solid solutions of significantly higher concentrations than the equilibrium ones),
• temperature decrease and ease of interaction of phases during further treatments such as calcination, hydration, sorption, reduction, etc.,
• modification of the operation properties (formability, strength, texture, etc.),
• simplification of technologies by reducing a number of stages and aggregate costs.
Some examples of the potential of MCA in the preparation of nanosized catalysts are presented below.
Preparation of nanosized ferrite catalytic samples (Fe3−xMexO4, 0 ≤ x ≤ 1, Me = Cu, Ni, Co, Mg, etc.) or their precursors in planetary ball mills leads to improvement of their catalytic properties in the deep oxidation of volatile organic compounds and methanol degradation.783a–c Ca and Cu ferrites, mechanochemically synthesized and thermally treated at low temperatures (450–500 °C), are highly active catalysts in the water-gas shift reaction.783d New intermetallic hydrides Mg2MHx (where M = Co, Fe and 5 ≤ x ≤ 6) are synthesized by MCA. Firstly, alloys are produced and then a hydrogenation process takes place. These hydrides are efficient catalysts for hydrogenation of acetylene and diene hydrocarbons to mono-olefins with selectivity close to 100%. MCA of the metal powders at hydrogen pressure of 100 atm has led to the synthesis of two previously unknown intermetallic hydrides: Mg2NiH6 (stable at ambient temperature) and MgCuH2, both exhibiting hydrogenation activity.777a,781a,b MCA is a promising alternative for the preparation of promoted heterogeneous catalysts. The activity and selectivity towards methanol synthesis of mechanochemically prepared supported Cu/ZnO catalysts show a strong dependence on milling atmosphere and equipment.784 V2O5, V2O5–TiO2, and VPO–Bi systems were the subject of mechanochemical treatment.785 The catalytic activity and selectivity in the oxidation of butane and benzene to maleic anhydride and o-xylene to phthalic anhydride were determined as a function of time of mechanochemical treatment. VTiO catalyst for production of phthalic anhydride from o-xylene can be synthesized by mechanochemical treatment of V2O5 and TiO2 mixture. The presence of nanosized V2O5 species provides the activity of this catalyst equal to industrial samples. Some of the samples operate at lower temperatures (70–100 °C) compared to industrial entities.786 Mechanochemical treatment of V2O5–MoO3 oxide mixture (V/Mo = 70/30 at%) has been performed under different conditions. This resulted in an increase of sample activity in n-butane and benzene oxidation reactions and an increase of the selectivity in maleic anhydride formation.787 Layered double hydroxide compounds are widely used as precursors for complex oxides containing M(II) and M(III) metals, acid–base catalysts, sorbents, and drugs as well as anion exchange matrices of the “anionic clay” type.781a,b,788 It has been demonstrated that the formation of magnesium-aluminum double hydroxides [Mg(1−x)Alx(OH)2]x+(An−x/n)·mH2O, (A = Cl−, NO3−, SO42−) occurs during mechanical activation of a mixture of magnesium hydroxide (brucite) with a chloride, nitrate or sulphate salt of aluminum in high-energy planetary-type activators. Amorphous organic–inorganic hybrid materials are of interest due to their application in recoverable immobilized catalysts. Wang et al. reported a novel solvent-free mechanochemical approach to synthesis of a catalytically active palladium(II)–phosphine coordination network and to synthesis of metal complexes. Shortening of the reaction time together with solvent-free operation makes it a promising preparative method for organic–inorganic hybrid materials.789 2 wt% Ag/Al2O3 catalysts have been prepared by solvent-free mechanochemical treatment using a ball mill. A remarkable increase of activity was shown in the reduction of nitrogen oxides with octane by lowering the light off temperature up to 150 °C compared with the same catalyst synthesized by wet impregnation. The best catalyst prepared from silver oxide showed 50% NOx conversion at 240 °C and 99% at 360 °C.790 It was shown781a,b that MCA is an efficient technological tool to increase catalyst strength. In the case of phosphate dehydrogenation catalyst, it is possible to increase the crushing strength of pellets by 1.5–2 times with preservation of the optimal pore structure. Mechanical forces can be applied in homogeneous catalysis as a new method to change the coordination sphere of transition metal complexes and to achieve higher selectivity and activity in transition metal-catalyzed reactions. The use of ultrasound has been considered as a method for selectively breaking the coordination bonds in coordination polymers and as a tool to facilitate ligands dissociation.791
Catalytic reactions can take place at the surface of previously mechanically activated catalysts without continuous input of mechanical energy. Thus, mechanical treatment of solids with low or even no activity leads to an increase of their catalytic activity. Silica, alumina and alumino-silicates can be activated to be valuable catalysts with the incorporation of small amounts of metals.778b
German scientists78,792 tested the model reaction of benzene hydrogenation for catalytic activity of mechanically activated Ni and Co powders
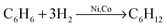 | (19) |
The dependence of benzene conversion on reaction temperature is shown in Fig. 48. There is no conversion to hexane for non-activated Ni-catalyst (0′) below 160 °C. At 220 °C the conversion is only 10%. The activity of the catalyst above this temperature goes down. However, all the activated Ni-catalysts (milling time 10–140 min) show much higher conversion for reaction (19), which proceeds by much lower temperatures in this case. Moreover, the interval of catalytic activity is broader in comparison with non-activated Ni. Eckell793 used nickel sheets to catalyze the hydrogenation of ethylene. He found that the activity was small before deformation, but increased significantly after cold rolling. This work was done before the concept of dislocation was widely accepted and it was not until more than 20 years later that Cratty and Granato794 interpreted Eckell’s results as evidence that emergent dislocations are active centers in heterogeneous catalysis. However, it is noteworthy to point out that Eckell attributed the change in activity of nickel because of cold working to a modification of the sliding properties of the metal. Thus, he intuitively introduced the dislocation concept years before these defects were discovered. Krause and Herman795 and Keating et al.796 observed that the deformation of platinum foils dramatically increased their catalytic activity toward dehydrogenation reactions. Uhara et al.797 studied the catalytic activity towards the dehydrogenation of formic acid on a series of wires of Ag, Au, Pt, Pd and Ni that were previously cold-worked by twisting. They observed that twisting of the metals dramatically increased their catalytic activity, which rapidly declined after annealing the wires at temperatures at which the recrystallization of the materials took place. The authors attributed the increase in catalytic activity to surface dislocations and vacancies, which were produced by cold working. The results summarized by Kishimoto798 are included in Fig. 49. This figure shows that the variation of the activity as a function of the annealing treatment would be considerably higher than the difference existing because of metal nature. This behavior suggests that proper mechanical treatment of non-noble metals like nickel could allow the substitution of noble metals like palladium and platinum as oxidation catalysts of CO in automobile industry. This shows the possible application as catalytic cartridge, because with the utilization of these materials it could operate at temperatures generally much lower than the recrystallization one. A correlation between chemical reactivity and deformation induced either by milling of nickel799 or by cold working by rolling of austenitic Fe–Cr–Ni has been reported elsewhere.800
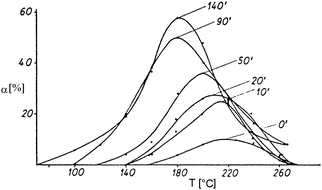 | ||
| Fig. 48 Benzene C6H6 conversion, α as a function of reaction temperature T, parameter: milling time (min)792a | ||
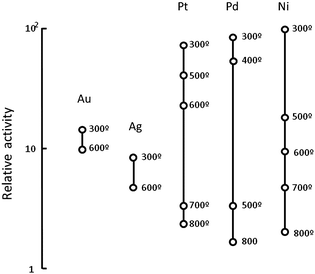 | ||
| Fig. 49 Comparison of catalytic activities of different metals. The solid lines represent the range of the change in the activity obtained at 200 °C. The figure designates the previous annealing temperature.798 | ||
The in situ synthesis of a copper-based nanocomposite powder with alumina is a consequence of aluminothermic reduction of CuO to Cu with Al2O3 formation.801–803 The metallic copper in Al2O3 nanoparticles is an active catalyst for hydrogen production in steam reforming of methanol. Metallic Ni or Cu catalysts supported on boehmite and alumina have been prepared by planetary ball milling under different conditions (wet or dry milling). These systems were tested in steam reforming of lower alcohols (methanol and ethanol) in the temperature range of 250–500 °C.804 The mechanical treatment of CuO with Al and Zn as well as Al and Mg metallic powders brings about the formation of metallic Cu with CuAl2O4–ZnAl2O4 and metallic Cu with CuAl2O4–MgAl2O4 double spinels, respectively. Catalytic properties of these materials were tested in the methanol steam reforming process. The results show that both Cu-double spinels are active towards hydrogen in the methanol steam reforming at 400 °C.805,806
Mechanochemical synthesis and mechanical activation of bismuth-doped vanadium-phosphorus oxide catalysts cause an improvement of their activity and selectivity in the reaction of partial oxidation of n-butane to maleic anhydride. An increase of about 40% in the total oxygen species is registered for a sample treated in ethanol. High amount of active oxygen released from V4+ phase (O–V4+ pair) was found to significantly contribute to the improvement of the catalytic properties.807 Hydrogen bond-driven self-assembly and mechanochemistry are used to facilitate supramolecular catalysis in the solid state. Mortar-and-pestle grinding proves to be an efficient means to achieve co-crystal formation and turnover using a physical mixture composed of an olefin and catalytic amounts of a ditopic template.808
Friščić et al.809a studied mechanochemical production of metal–organic framework ZIF-8 (sold as Basolite Z1200) from the simplest and non-toxic components. Materials such as ZIF-8 are rapidly gaining popularity for capturing large amounts of CO2 and, if manufactured cheaply and sustainably, could become widely used for carbon capture, catalysis and even hydrogen storage. Unprecedented methodology was used for real-time observation of reaction kinetics, reaction intermediates and to detect the changes as they happened. The high-energy X-ray beam from synchrotron radiation was used to get inside the jar and to probe the mechanochemical formation of ZIF-8. This technique can be applied for investigation and optimization of industrial processing of all types of chemical reactions in a ball mill.
The copper(I)-catalysed azide–alkyne cycloaddition (the CuAAC ‘click’ reaction) is proving to be a powerful new tool for the construction of mechanically interlocked molecular-level architectures.809b A multi-component CuAAC reaction can be conducted using mechanochemical treatment. The first copper vial catalysed CuAAC reaction was reported in ref. 809c. Under solvent-free mechanochemical conditions the reaction is complete in as little as 15 minutes and the triazole product isolated straight from the reaction vial with no further purification.
An elegant synthesis of 3-carboxycoumarins (an important class of biologically active compounds) without using a solvent and with grinding of the reagents was proposed in ref. 809d. In the second step of the reaction, sodium or ammonium acetate plays the role of catalyst. The target product, the yield of which approaches 100%, is isolated extremely simply: the catalyst is washed with water. Conventional methods for synthesis of 3-carboxycoumarins require the use of dimethylformamide, POCl3, piperidine as the base, and result in fairly small yields of the target product.
Mechanochemical approaches, in which co-grinding of reactants and catalytic additives results in the self-assembly of advanced materials, such as porous metal–organic frameworks (MOFs), directly from the simplest and cheapest possible precursors are presented in ref. 809e,f. These advanced mechanochemical methodologies include liquid-assisted grinding (LAG) and ion- and liquid-assisted grinding (ILAG). The methodologies allow chemical reactions to be conducted independently of the solubility of the reactant materials and lead to overall 1000- or 10000-fold reductions in the use of solvent and energy compared to conventional processes.
When MCA is combined with a catalytic process, a special pathway for the process appears. Under these conditions, fresh surfaces with broken and distorted bonds with radical active sites are continuously generated. The appearance of excited atoms on such a surface determines their reactivity. The lifetime of these atoms is comparable with the chain termination rate. In this case, the reaction with a gas goes without any activation barrier. Radical reactions are defined by the rates of formation and consumption of the active sites that are proportional to the reproduction rate of fresh surfaces. The sites are consumed by spontaneous annihilation and interaction with gases during the catalytic act.777 Mechanically activated reactions of organic compound synthesis are of two types: stoichiometric (when the reagents are directly mechanically activated) and catalytic (the catalyst is activated). Examples of the use of stoichiometric mechanically activated processes in fine-particle organic synthesis have been reported.777 Blair et al. developed novel catalysts specifically designed for mechanocatalysis. They have found that cellulose can be mechanocatalytically broken down into saccharide dehydration products. Under the correct conditions, 80% of the available cellulose can be converted to simple sugars. This approach will reduce the dependence on starch-based sources such as corn for production of ethanol and biomass-derived chemicals.810
The use of scanning probe microscopy-based techniques (AFM) to manipulate single molecules and deliver them in a precisely controlled manner to a specific target represents a novel nanotechnological challenge in catalyst preparation.811
8.3 Extractive metallurgy
According to the classical view, extractive metallurgy is the art and science of extracting metals from their ores by chemical methods.812 It is actually divided into three sectors: hydrometallurgy, pyrometallurgy and electrometallurgy. Hydrometallurgy is the technology of extracting metals from ores by aqueous methods, pyrometallurgy by dry thermal methods, and electrometallurgy by electrolytic methods.The ACTIVOX™ process was developed in Australia as an alternative to the pretreatments of sulphidic concentrates by roasting and bacterial oxidation.813 The process has been applied to the recovery of non-ferrous and precious metals from concentrates and calcines. The principle of the process is shown in Fig. 50. ACTIVOX™ is a hydrometallurgical process combining ultra-fine milling to a P80 of 10 μm with a low-temperature (100 °C), low-pressure (1000 kPa) oxidative leach to liberate metals from a sulphide matrix. Base metals (i.e. copper, zinc, nickel and cobalt) are extracted into the leach liquor, while gold and silver remain in the leach residue in a form suitable for further processing. The working conditions favour the formation of elemental sulphur over sulphate, thereby using less oxygen (usually less than 1.5 kg of O2 per kg of S) than is required for complete oxidation to sulphate (typically 2.2 kg O2 per kg S). Other features include short oxidation times (1–2 hours), clean pregnant liquors, and the possibility of treating environmentally hazardous species such as arsenopyrite to produce stable ferric arsenate residues.814 As for activation equipment, Netzsch-IsaMill with capacity 10000 L was tested (Fig. 51).
 | ||
| Fig. 50 ACTIVOX™ process.813d | ||
 | ||
| Fig. 51 The horizontal stirred mill – IsaMill.815 | ||
The MELT process was developed in Slovakia for hydrometallurgical treatment of tetrahedrite sulphidic concentrates in order to obtain antimony in a soluble form and copper as copper sulphide.816–818 The process name is an abbreviation: MEchanochemical Leaching of Tetrahedrites. The chemistry of the reaction between tetrahedrite (simplified formula Cu3SbS3) and Na2S can be described in a simplified form by the equations819
| 2Cu3SbS3 + Na2S → 3Cu2S + 2NaSbS2 | (20) |
| NaSbS2 + Na2S → Na3SbS3 | (21) |
The soluble Na3SbS3 containing trivalent antimony is oxidized to a product containing pentavalent antimony by the polysulphide ions present in the leaching liquor
| (x − 1)Na3SbS3 + Na2Sx → (x − 1)Na3SbS4 + Na2S | (22) |
The principal flowsheet of operation, in which mechanochemical leaching(I) is followed by chemical leaching(II), is shown in Fig. 52. Alkaline solutions of Na2S have been explored as leaching agent. The documented results show that almost total extraction of Sb can be achieved by alkaline Na2S leaching at 95 °C leaving 0.25% Sb in solid residue. The MELT process was developed and verified in a laboratory attritor822 and in semi-industrial attritor.6,820,821 It was further tested in a pilot plant hydrometallurgical unit in Rudňany, Slovakia (Fig. 53). Fig. 54 summarizes the flowsheet of this process. Tests have shown that alkaline Na2S leaching for 30 min at 86 °C and atmospheric pressure are required to achieve total extraction of antimony. The other valuable metals (Cu, Ag, Au) form the main economic components of solid residue. The liquid/solid ratio (2.5–4.8) and power inputs (150–250 kW h t−1) that were applied in optimized mechanochemical experiments are acceptable for plant operation.817
 | ||
| Fig. 52 Flowsheet of leaching unit: 1 – heating, 2 – chemical reactor, 3 – pump, 4 – valve, 5 – attritor, 6 – cooling. Working regimes: I – mechanochemical leaching, II – chemical leaching. Reprinted with permission from ref. 6. Copyright 2000, Elsevier. | ||
 | ||
| Fig. 53 Pilot plant unit at Rudňany (Slovakia): unit and LME Netzsch attritor in combination with chemical reactor.821 | ||
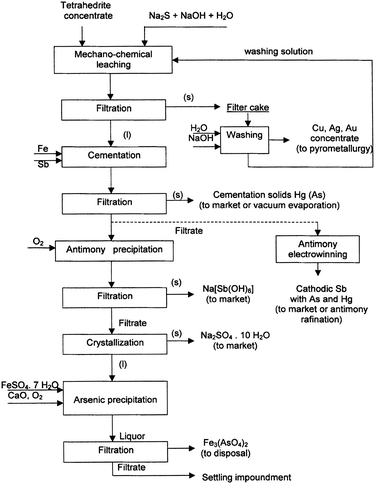 | ||
| Fig. 54 Flowsheet of the MELT process. Reprinted with permission from ref. 818. Copyright 2006, Elsevier. | ||
9. Conclusions and outlook
Mechanochemistry has made significant progress during the last ten years. The available theoretical, experimental and applied results are summarized in this review article.The identified hallmarks of mechanochemistry include:
• influencing reactivity by creating interphases (especially in composite and multi/phase systems), defects in solids and by existence of relaxation phenomena,
• creating well-crystalized cores of nanoparticles with disordered near-surface shell regions,
• performing simple, dry, time-convenient one-step syntheses,
• preparing nanomaterials with properties set in advance,
• scaling up to industrial production, and
• enabling work under environmentally friendly and essentially waste-free conditions.
In general, there is still a long way to go in mechanochemistry. Mainly the elucidation of the mechanism of the reactions725a and industrial application of mechanochemistry7 in nanotechnology are the issues. However, there is no doubt that mechanochemistry should be incorporated in textbooks of not only solid-state, but also other branches of chemistry. The same idea was proposed by Wilhelm Ostwald (Nobel Prize) more than one hundred years ago.
Abbrevations
| BM | Ball milling |
| BPR | Ball-to-powder ratio |
| MA | Mechanical activation, mechanical alloying |
| MAFAPAS | Mechanically activated field-activated pressure-assisted synthesis |
| MARES | Mechanically activated reactive extrusion synthesis |
| MARFOS | Mechanically activated reactive forging synthesis |
| MASHS | Mechanically activated self-heat-sustaining reaction |
| MASPS | Mechanically activated spark plasma synthesis |
| MCA | Mechanochemical activation |
| MCS | Mechanochemical synthesis |
| MSR | Mechanically induced self-sustaining reaction |
| PCA | Process control agent |
| SSM | Solid-state metathesis reaction |
| SHS | Self-heat-sustaining reaction, self-propagating high-temperature synthesis |
Acknowledgements
The authors acknowledge support of the following institutions (and grants): the Slovak Grant Agency VEGA (2/0009/11: P. Baláž, MB, ED; 2/0043/11: MA), the Slovak Agency for Science and Development APVV (VV-0189-10: P. Baláž, MB, ED, MA; VV-0528-11: P. Billik), Centre of Excellence of Slovak Academy of Sciences (CFNT-MVEP), Slovenian Research Agency (TR), Russian Foundation for Basic Research (10-03-00942a: AS, 12-03-00651a: AS), Ministry of Science and Higher education in Poland (CUT/c-1/DS/KWC/2008-2012, PB1T09B02330, NN209145136, NN209148936: KW), Alexander von Humboldt Foundation and EPFL (MS). This acknowledgement is also devoted to all the colleagues who have contributed to this review article, but are not listed as co-authors. They are too numerous to be named individually, but they can be found in the cited publications. We are grateful that we got to know them and enjoyed the cooperation.Notes and references
- IUPAC Compendium of Chemical Technology, (the “Gold Book”), ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed
; XML on-line corrected version: http://goldbook.iupac.org, created by M. Nic, J. Jirat and B. Kosata, updates compiled by A. Jenkins, 2006.
-
(a) W. Ostwald, Lehrbuch der Allgemeinen Chemie, 1. Aufgabe, 2. Band, Leipzig, 1887 Search PubMed
- W. Ostwald, Die Welt der Vernachlässigten Dimensionen, Verlag von Theodor Steinkopff, Dresden, 1927 Search PubMed
-
(a) A. Smékal, Naturwissenschaften, 1942, 30, 224 CrossRef Search PubMed
-
(a) L. Takacs, J. Met., 2000, 12 CAS Search PubMed
- P. Baláž, Extractive Metallurgy of Activated Minerals, Elsevier, Amsterdam, 2000 Search PubMed
- P. Baláž, Mechanochemistry in Nanoscience and Minerals Engineering, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed
-
(a) I. J. Lin and S. Nadiv, Mater. Sci. Eng., 1970, 393, 193 Search PubMed
-
(a) F. P. Bowden and A. Yoffe, Initiation and Growth of Explosion in Liquids and Solids, Cambridge University Press, Cambridge, 1952 Search PubMed
- P. A. Thiessen, K. Meyer and G. Heinicke, Grundlagen der Tribochemie, Akademie-Verlag, Berlin, 1967 Search PubMed
-
(a) E. M. Gutman, Mechanochemistry of Metals and Protection against Corrosion, Metallurgiya, Moscow, 1974 (in Russian) Search PubMed
- D. L. Zhang, Prog. Mater. Sci., 2004, 49, 537 CrossRef CAS Search PubMed
- S. Rosenkranz, S. Breitung-Faes and A. Kwade, Powder Technol., 2011, 212, 224 CrossRef CAS Search PubMed
- C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1 CrossRef CAS Search PubMed
- P. R. Sooni, Mechanical Alloying: Fundamentals and Applications, Cambridge International Science Publishing, Cambridge, 2001 Search PubMed
- E. Avvakumov, M. Senna and N. Kostova, Soft Mechanochemical Synthesis: A Basis for New Chemical Technologies, Kluwer Academic Publischers, Dordrecht, 2001 Search PubMed
- P. Billik and M. Čaplovičová, in Advances in Nanotechnology, ed. Z. Bartul and J. Trenor, Nova Publishers, New York, 2012, vol. 8, pp. 111–164 Search PubMed
- H. Heegn, F. Birkender and A. Kamptner, Cryst. Res. Technol., 2003, 38, 7 CrossRef CAS Search PubMed
- E. L. Fokina, N. I. Budim, V. G. Kochnev and G. G. Chernik, J. Mater. Sci., 2004, 39, 5217 CrossRef CAS Search PubMed
- P. P. Chattopadhyay, I. Manna, S. Talapatra and S. K. Pabi, Mater. Chem. Phys., 2001, 68, 85 CrossRef CAS Search PubMed
- J. Alkebro, S. Bégin-Colin, A. Mocellin and R. Warren, J. Solid State Chem., 2002, 164, 88 CrossRef CAS Search PubMed
- K. S. Venkataraman and K. S. Narayanan, Powder Technol., 1998, 96, 190 CrossRef CAS Search PubMed
- L. Lu and M. O. Lai, Mechanical Alloying, Kluwer Academic Publishers, Boston, 1998 Search PubMed
- C. Suryanarayana, Mechanical Alloying and Milling, Marcel Dekker, New York, 2004 Search PubMed
- L. Takacs, Prog. Mater. Sci., 2002, 47, 355 CrossRef CAS Search PubMed
- Y. Ogino, T. Yamasaki, M. Miki, N. Atsumi and K. Yoshioka, Scr. Metall. Mater., 1993, 28, 967 CrossRef CAS Search PubMed
- Z. H. Chin and T. P. Perng, Appl. Phys. Lett., 1997, 70, 2380 CrossRef CAS Search PubMed
- G. Mulas, L. Schiffini, G. Tanda and G. Cocco, J. Alloys Compd., 2005, 404–406, 343 CrossRef CAS Search PubMed
- F. Delogu and G. Mulas, Int. J. Hydrogen Energy, 2009, 34, 3026 CrossRef CAS Search PubMed
- M. D. Chio, L. Schiffini, S. Enzo, G. Cocco and M. Baricco, Renewable Energy, 2008, 33, 237 CrossRef Search PubMed
- G. Mulas, L. Schiffini and G. Cocco, J. Mater. Res., 2004, 19, 3279 CrossRef CAS Search PubMed
- P. Unifantowicz, Z. Oksiuta, P. Olier, Y. de Carlan and N. Baluc, Fusion Eng. Des., 2011, 86, 2413 CrossRef CAS Search PubMed
- M. I. Flores-Zamora, C. A. Martinez-Pérez, M. Garcia-Guaderrama, I. Estrada-Guel, F. Espinoza-Magana and R. Martínez-Sánchéz, Rev. Adv. Mater. Sci., 2008, 18, 301 CAS Search PubMed
- A. Morales-Rodriguez, A. Gallardo-López, A. Dominguez-Rodriguez, J. M. Córdoba, M. A. Avilés and F. J. Gotor, J. Eur. Ceram. Soc., 2011, 31, 299 CrossRef CAS Search PubMed
- E. Chicardi, J. M. Córdoba, M. J. Sayagués and F. J. Gotor, Int. J. Refract. Met. Hard Mater., 2012, 31, 39 CrossRef CAS Search PubMed
- L. Takacs and J. S. McHenry, J. Mater. Sci., 2006, 41, 5246 CrossRef CAS Search PubMed
- M. Abdellaoui and E. Gaffet, Acta Mater., 1996, 44, 725 CrossRef CAS Search PubMed
- R. M. Davis and C. C. Koch, Scr. Metall., 1987, 21, 305 CrossRef CAS Search PubMed
-
(a) L. Opoczky, Powder Technol., 1977, 17, 1 CrossRef CAS Search PubMed
- E. M. Gutman, Mechanochemistry of Materials, Cambridge International Science Publishing, Cambridge, 1998 Search PubMed
- V. I. Levitas, in High Pressure Surface Science and Engineering, ed. Y. Gogotsi and V. Domnich, Institute of Physics, Bristol, 2004, pp. 159–292 Search PubMed
- L. Onsager, Phys. Rev., 1931, 37, 405 CrossRef CAS Search PubMed
- S. R. Berry, S. A. Rice and J. Ross, Matter in Equilibrium: Statistical Mechanics and Thermodynamics, Cambridge University Press, Cambridge, 2nd edn, 2004 Search PubMed
- Physical Metallurgy, ed. R. W. Cahn and P. Haasen, Elsevier, Amsterdam, 4th edn, 1996 Search PubMed
- P. Bellon and R. S. Averback, Phys. Rev. Lett., 1995, 74, 1819 CrossRef CAS Search PubMed
- J. E. Hammerberg, B. L. Holian, J. Röder, A. R. Bishop and S. J. Zhou, Phys. D, 1998, 123, 330 CrossRef CAS Search PubMed
- X.-J. Fu, M. L. Falk and D. A. Rigney, Wear, 2001, 250, 420–430 CrossRef Search PubMed
- A. C. Lund and C. A. Schuh, Appl. Phys. Lett., 2003, 82, 2017 CrossRef CAS Search PubMed
- A. C. Lund and C. A. Schuh, J. Appl. Phys., 2004, 95, 4815 CrossRef CAS Search PubMed
- F. Delogu and G. Cocco, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 144108 CrossRef Search PubMed
- F. Delogu and G. Cocco, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 014124 CrossRef Search PubMed
- S. Odunuga, Y. Li, P. Krasnochtchekov, P. Bellon and R. S. Averback, Phys. Rev. Lett., 2005, 95, 045901 CrossRef CAS Search PubMed
- F. Delogu, J. Appl. Phys., 2008, 104, 073533 CrossRef Search PubMed
- N. Q. Vo, S. Odunuga, P. Bellon and R. S. Averback, Acta Mater., 2009, 57, 3012 CrossRef CAS Search PubMed
- Y. Ashkenazy, N. Q. Vo, D. Schwen, R. S. Averback and P. Bellon, Acta Mater., 2012, 60, 984 CrossRef CAS Search PubMed
- F. Delogu and G. Cocco, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035406 CrossRef Search PubMed
- F. Delogu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 014115 CrossRef Search PubMed
- F. Delogu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205415 CrossRef Search PubMed
- F. Delogu, Chem. Phys. Lett., 2012, 521, 125 CrossRef CAS Search PubMed
- D. A. Rigney, Wear, 2000, 245, 1 CrossRef CAS Search PubMed
- J. Song and D. Srolovitz, Acta Mater., 2006, 54, 5305 CrossRef CAS Search PubMed
- J. Song and D. Srolovitz, J. Appl. Phys., 2008, 104, 124312 CrossRef Search PubMed
- A. Fortini, M. I. Mendelev, S. Buldyrev and D. Srolovitz, J. Appl. Phys., 2008, 104, 074320 CrossRef Search PubMed
- H. Kim and A. Strachan, Phys. Rev. Lett., 2010, 104, 215504 CrossRef Search PubMed
- H. Kim and A. Strachan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 024108 CrossRef Search PubMed
- F. Delogu, J. Appl. Phys., 2011, 110, 103505 CrossRef Search PubMed
- M. Nosonovsky and B. Bhushan, Multiscale Dissipative Mechanisms and Hierarchical Surfaces, Springer-Verlag, Berlin, 2008 Search PubMed
- R. Chattopadhyay, Surface Wear. Analysis, Treatment, and Prevention, ASM International, Materials Park, 2001 Search PubMed
- D. A. Rigney and J. E. Hammerberg, in The 1999 Julia R. Weertman Symposium on Advanced Materials for the 21st Century, ed. Y. W. Chung, D. C. Dunand, P. K. Liaw and G. B. Olson, TMS (OH), Cincinnati, 1999, pp. 465–474 Search PubMed
- H.-J. Kim, W. K. Kim, M. J. Falk and D. A. Rigney, Tribol. Lett., 2007, 28, 299 CrossRef Search PubMed
- H.-J. Kim, S. Karthikeyan and D. A. Rigney, Wear, 2009, 267, 1130 CrossRef CAS Search PubMed
- S. Karthikeyan, A. Agrawal and D. A. Rigney, Wear, 2009, 267, 1166 CrossRef CAS Search PubMed
- A. Emge, S. Karthikeyan and D. A. Rigney, Wear, 2009, 267, 562 CrossRef CAS Search PubMed
- D. Rigney and S. Karthikeyan, Tribol. Lett., 2009, 39, 3 CrossRef Search PubMed
- Abrasion Resistance of Materials, ed. M. Adamiak, InTech, Rijeka, 2012 Search PubMed
- R. W. Hertzberg, Deformation and Fracture Mechanics of Engineering Materials, John Wiley & Sons, New York, 1995 Search PubMed
- Fracture of Nano and Engineering Materials and Structures, ed. E. Gdoutos, Springer, Dordrecht, 2006 Search PubMed
- G. Heinicke, Tribochemistry, C. H. Verlag, Munich, 1985 Search PubMed
- T. E. Fisher, Annu. Rev. Mater. Sci., 1988, 18, 303 CrossRef Search PubMed
- J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, VCH Publishers, New York, 2005 Search PubMed
- P. Meloni, G. Carcangiu and F. Delogu, Mater. Res. Bull., 2012, 47, 146 CrossRef CAS Search PubMed
- J. A. Harrison and D. W. Brenner, J. Am. Chem. Soc., 1994, 116, 10399 CrossRef CAS Search PubMed
- S. M. Hsu, J. Zhang and Z. Yin, Tribol. Lett., 2002, 13, 131 CrossRef CAS Search PubMed
- C. L. Rountree, R. K. Kalia, E. Lidorikis, A. Nakano, L. Van Brutzel and P. Vashishta, Annu. Rev. Mater. Res., 2002, 32, 377 CrossRef CAS Search PubMed
- K. Muralidharan, J. H. Simmons, J. H. Deymier and K. Runge, J. Non-Cryst. Solids, 2005, 351, 1532 CrossRef CAS Search PubMed
- M. J. Buehler, Atomistic Modeling of Materials Failure, Springer, New York, 2008 Search PubMed
- F. Musso, P. Ugliengo, X. Solans-Monfort and M. J. Sodupe, J. Phys. Chem. C, 2010, 114, 16430 CAS Search PubMed
- S. Giordano, A. Mattoni and L. Colombo, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz, John Wiley & Sons, New York, 2011, pp. 1–83 Search PubMed
- F. Delogu, J. Phys. Chem. C, 2011, 115, 21230 CAS Search PubMed
- V. V. Boldyrev, Experimental Methods in the Mechanochemistry of Inorganic Solids, Nauka, Novosibirsk, 1983 (in Russian) Search PubMed
- E. G. Avvakumov, Mechanical Methods for Activation of Chemical Processes, Nauka, Novosibirsk, 1986 (in Russian) Search PubMed
- P. Yu. Butyagin, Usp. Khim., 1994, 63, 1031 CAS Search PubMed
- Advances in Mechanochemistry: Physical and Chemical Processes under Deformation, ed. P. Yu. Butyagin and A. Dubinskaya, Harwood Academic Publishers, Amsterdam, 1998 Search PubMed
- F. Kh. Urakaev and V. V. Boldyrev, Powder Technol., 2000, 107, 93 CrossRef CAS Search PubMed
- F. Kh. Urakaev and V. V. Boldyrev, Powder Technol., 2000, 107, 197 CrossRef CAS Search PubMed
- I. F. Vasconcelos and R. S. de Figuereido, J. Phys. Chem. B, 2003, 107, 3761 CrossRef CAS Search PubMed
- P. L. Houston, Chemical Kinetics and Reaction Dynamics, Dover, New York, 2006 Search PubMed
- G. S. Chodakov, Physics of Milling, Nauka, Moscow, 1972 (in Russian) Search PubMed
- V. Klika and F. Maršík, J. Phys. Chem. B, 2009, 113, 14689 CrossRef CAS Search PubMed
- V. I. Levitas, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 184118 CrossRef Search PubMed
- P. Yu. Butyagin, Russ. Chem. Rev., 1971, 40, 901 CrossRef Search PubMed
- V. I. Levitas, Y. Ma, J. Hashemi, M. Holtz and N. Guven, J. Chem. Phys., 2006, 125, 044507 CrossRef Search PubMed
- V. I. Levitas and O. M. Zarechnyy, Appl. Phys. Lett., 2007, 91, 141919 CrossRef Search PubMed
- V. I. Levitas and O. M. Zarechnyy, Europhys. Lett., 2009, 88, 16004 CrossRef Search PubMed
- V. I. Levitas, Y. Ma, E. Selvi, J. Wu and J. A. Patten, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054114 CrossRef Search PubMed
- A. P. Zhilyaev and T. G. Langdon, Prog. Mater. Sci., 2006, 53, 893 CrossRef Search PubMed
- F. Delogu, Scr. Mater., 2012, 67, 340 CrossRef CAS Search PubMed
- F. Delogu, Acta Mater., 2008, 56, 905 CrossRef CAS Search PubMed
- Experimental and Theoretical Studies in Modern Mechanochemistry, ed. F. Delogu and G. Mulas, Transworld Research Network, Trivandrum, 2010 Search PubMed
- High-Energy Ball Milling. Mechanochemical Processing of Nanopowders, ed. M. Sopicka-Lizer, Woodhead Publishing, Cambridge, 2010 Search PubMed
- B. Leory, Am. J. Phys., 1985, 53, 346 CrossRef Search PubMed
- F. Delogu and G. Cocco, J. Alloys Compd., 2007, 436, 233 CrossRef CAS Search PubMed
- F. Delogu, Scr. Mater., 2008, 58, 126 CrossRef CAS Search PubMed
- F. Delogu, Acta
Mater., 2008, 56, 2344 CrossRef CAS Search PubMed
- F. Delogu, Acta Mater., 2011, 59, 2069 CrossRef CAS Search PubMed
- M. J. Buerger, Phase Transformations in Solids, John Wiley and Sons, New York, 1951 Search PubMed
- C. N. R. Rao and K. J. Rao, Phase Transitions in Solids, McGraw-Hill, New York, 1978 Search PubMed
- G. L. Clark and R. Rowan, J. Am. Chem. Soc., 1941, 63, 1302 CrossRef CAS Search PubMed
- F. Dachille and R. Roi, Nature, 1960, 186, 34 CrossRef CAS Search PubMed
- D. Lewis, D. O. Northwood and R. C. Reeve, J. Appl. Crystallogr., 1969, 2, 156 CrossRef CAS Search PubMed
- I. J. Lin and S. Niedzwiedz, J. Am. Ceram. Soc., 1973, 56, 62 CrossRef CAS Search PubMed
- M. Senna and H. Kuno, J. Am. Ceram. Soc., 1971, 54, 259 CrossRef CAS Search PubMed
- M. Senna, Cryst. Res. Technol., 1985, 20, 209 CrossRef CAS Search PubMed
- J. H. Burns and M. A. Breding, J. Chem. Phys., 1956, 25, 1281 CrossRef CAS Search PubMed
- J. C. Jamieson and J. R. Goldsmith, Am. Mineral., 1960, 45, 818 CAS Search PubMed
- N. Kawashima and K. Meguro, Bull. Chem. Soc. Jpn., 1975, 48, 1857 CrossRef CAS Search PubMed
- J. M. Criado and J. M. Trillo, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 961 RSC
- G. Martínez, J. Morales and G. Munuera, J. Colloid Interface Sci., 1980, 81, 500 CrossRef Search PubMed
- R. B. Gammage and D. R. Glasson, J. Colloid Interface Sci., 1976, 55, 396 CrossRef CAS Search PubMed
- T. Isobe and M. Senna, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1199 RSC
- Y. Iguchi and M. Senna, Powder Technol., 1985, 43, 155 CrossRef CAS Search PubMed
-
(a) J. M. Criado, M. J. Diánez and J. Morales, J. Mater. Sci., 2004, 39, 5189 CrossRef CAS Search PubMed
- I. J. Lin and P. Somasundaran, Powder Technol., 1972, 6, 171 CrossRef CAS Search PubMed
- J. J. Lin, S. Nadiv and D. J. M. Grodzian, Miner. Sci. Eng., 1975, 7, 313 Search PubMed
- K. A. Gross, Philos. Mag., 1965, 12, 805 CrossRef Search PubMed
- R. Schrader and B. Hoffmann, Z. Anorg. Allg. Chem., 1969, 369, 41 CrossRef CAS Search PubMed
- P. G. Fox and J. Soria-Ruiz, Proc. R. Soc., 1970, A314, 429 CrossRef Search PubMed
- A. W. Momber, J. Mater. Sci., 2010, 45, 750 CrossRef CAS Search PubMed
- J. M. Criado, F. González and J. Morales, Thermochim. Acta, 1979, 32, 99 CrossRef CAS Search PubMed
- G. Martinez, J. Morales and G. Munuera, J. Colloid Interface Sci., 1981, 81, 500 CrossRef CAS Search PubMed
- J. M. Criado and C. Real, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2765 RSC
- J. M. Criado, C. Real and J. Soria, Solid State Ionics, 1989, 32–33, 461 CrossRef Search PubMed
- S. Begin-Colin, T. Girot, G. Le Cäer and A. Mocellin, J. Solid State Chem., 2000, 149, 41 CrossRef CAS Search PubMed
- X. Pan and M. Xueming, J. Solid State Chem., 2004, 177, 4098 CrossRef CAS Search PubMed
- S. Begin-Colin, A. Gadalla, G. Le Cäer, O. Humbert, F. Thomas, G. Barses, F. Villieras, L. F. Toma, G. Bertrand, O. Zahraa, M. Gallart, B. Honerlage and P. Gilliot, J. Phys. Chem. C, 2009, 113, 16589 CAS Search PubMed
- E. Arca, G. Mulas, F. Delogu, J. Rodriguez-Ruiz and S. Palmas, J. Alloys Compd., 2009, 477, 583 CrossRef CAS Search PubMed
- J. Šubrt, J. M. Criado, L. Szatmary, M. J. Diánez, N. Murafa, L. Pérez-Maqueda and V. Brezová, Int. J. Photoenergy, 2011, 156941 Search PubMed
- S. Palmas, A. M. Polcaro, J. Rodriguez-Ruiz, A. Da Pozzo, A. Vacca, M. Mascia, F. Delogu and P. C. Ricci, Int. J. Hydrogen Energy, 2009, 34, 9662 CrossRef CAS Search PubMed
- F. Delogu, J. Alloys Compd., 2009, 468, 22 CrossRef CAS Search PubMed
- G. Liu, X. Wang, Z. Chen, H. M. Cheng, G. Qing and M. Lu, J. Colloid Interface Sci., 2009, 329, 331 CrossRef CAS Search PubMed
- G. W. Lee, S. Y. Bsng, C. Lee, W. M. Kim, D. Kim, K. Kim and N. G. Park, Curr. Appl. Phys., 2009, 9, 900 CrossRef Search PubMed
- M. Rezaee and S. M. M. Khoie, J. Alloys Compd., 2010, 507, 484 CrossRef CAS Search PubMed
- T. P. Ang, J. Y. Law and Y. F. Han, Catal. Lett., 2010, 139, 77 CrossRef CAS Search PubMed
- M. Rezaee, S. M. M. Khoie and K. H. Liu, CrystEngComm, 2011, 13, 5055 RSC
- J. Li, S. Yin, Y. Wang and T. Sato, J. Catal., 2012, 286, 273 CrossRef Search PubMed
- M. Senna, N. Myers, A. Aimable, V. Laporte, C. Pulgarin, O. Baghriche and P. Bowen, J. Mater. Res., 2012, 28, 354 CrossRef Search PubMed
- S. Yin, Q. Zhang, F. Saito and T. Sato, in High Energy Ball Milling, ed. M. Sopicka-Lizer, CRC Press, Washington DC, 2010, pp. 304–330 Search PubMed
- J. Judes and V. Kamaraj, Mater. Sci.-Pol., 2009, 27, 407 CAS Search PubMed
- J. M. Criado, M. González, M. J. Diánez, L. A. Pérez-Maqueda and J. Málek, J. Phys. Chem. Solids, 2007, 68, 824 CrossRef CAS Search PubMed
- C. Frondel and C. Palache, Science, 1948, 107, 602 CAS Search PubMed
- H. Kuwamoto, J. Mater. Sci., 1985, 4, 940 CAS Search PubMed
- M. Farkas-Jahnke and P. Gács, Krist. Tech., 1979, 14, 1475 CrossRef CAS Search PubMed
- E. T. Allen, J. L. Crenshaw and H. E. Merwin, Am. J. Sci., 1912, 34, 341 CrossRef Search PubMed
- M. A. Schort and E. G. Steward, Z. Phys. Chem., 1957, 13, 298 CrossRef Search PubMed
- K. Imamura and M. Senna, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 1131 RSC
- M. Senna, in Proceedings of International Symposium on Powder Technology, Kyoto, 1981, pp. 457–464 Search PubMed
- P. Baláž, Z. Bastl, J. Briančin, I. Ebert and J. Lipka, J. Mater. Sci., 1992, 27, 653 CrossRef Search PubMed
-
(a) J. Y. Huang, H. Yasuda and H. Mori, J. Am. Ceram. Soc., 2000, 83, 403 CrossRef CAS Search PubMed
- A. N. Streletskii, D. G. Permenov, B. B. Bokhonov, I. V. Kolbanev, A. V. Leonov, I. V. Berestetskaya and K. A. Streletzky, J. Alloys Compd., 2009, 483, 313 CrossRef CAS Search PubMed
-
(a) G. Mestl, B. Herzog, R. Schlogl and H. Knozinger, Langmuir, 1995, 11, 3027 CrossRef CAS Search PubMed
- A. N. Streletskii, I. V. Kolbanev, A. Ju. Dolgoborodov and A. B. Borunava, Combustion and Explosion, ed. S. M. Frolov, Torus Press, Moscow, 2011, vol. 4, pp. 166–171 (in Russian) Search PubMed
-
(a) F. Salver-Disma, J.-M. Taraskon, C. Clinard and J.-N. Rouzaud, Carbon, 1999, 37, 1941 CrossRef CAS Search PubMed
- A. N. Streletskii, I. V. Kolbanev, A. B. Borunova and P. Yu. Butyagin, in Experimental and Theoretical Studies in Modern Mechanochemistry, ed. F. Delogu and G. Mulas, Transworld Research Network, Trivandrum, 2010, pp. 169–182 Search PubMed
-
(a) E. Gaffet and M. Harmelin, J. Less Common Met., 1990, 157, 201 CrossRef CAS Search PubMed
-
(a) A. N. Streletskii, A. V. Leonov, I. V. Berestetskaja, S. N. Mudretsova, A. F. Maiorova and P. Yu. Butyagin, J. Metastable Nanocryst. Mater., 2002, 13, 187 CrossRef Search PubMed
-
(a) A. N. Streletskii, I. V. Kolbanev, V. A. Radzig and A. B. Borunava, Programme and the Book of Abstracts of 13th Annual Conference Yucomat, Herceg Novi, 2011, p. 14 Search PubMed
- K. L. Da Silva, D. Menzel, A. Feldhoff, C. Kubel, M. Bruns, A. Paezano, A. Duvel, M. Wilkening, M. Ghafari, F. J. Litterst, P. Heitjans, K. Becker and V. Šepelák, J. Phys. Chem., 2011, 115, 7209 CAS Search PubMed
-
(a) D. Oleszak and P. H. Shingu, J. Appl. Phys., 1996, 79, 2975 CrossRef CAS Search PubMed
-
(a) H. J. Fecht, Nanostruct. Mater., 1995, 6, 33 CrossRef CAS Search PubMed
- L. S. Vasil’ev and I. L. Lomaev, Phys. Met. Metallogr., 2006, 101, 386 CrossRef Search PubMed
- J. Eckert, J. Holzer, C. E. Krill and W. L. Johnson, J. Mater. Res., 1992, 7, 1751 CrossRef CAS Search PubMed
- J. Eckert, Nanostruct. Mater., 1995, 6, 413 CrossRef CAS Search PubMed
- P. Yu. Butyagin, Dokl. Khim., 1993, 331, 311 CAS Search PubMed
- F. A. Mohamed, Acta Mater., 2003, 51, 4107 CrossRef CAS Search PubMed
-
(a) P. Yu. Butyagin, I. V. Berestetskaya, I. V. Kolbanev and I. K. Pavlyshev, Russ. J. Phys. Chem., 1986, LX, 343 Search PubMed
- R. A. Andrievski, Usp. Fiz. Nauk, 2007, 177, 721 CrossRef Search PubMed
- P. Wang, S. Orimo, T. Matsushima and H. Fujii, Appl. Phys. Lett., 2002, 80, 318 CrossRef CAS Search PubMed
- P. Yu. Butyagin, A. N. Streletskii, I. V. Berestetskaja and A. B. Borunava, Colloid J., 2001, 63, 639 CrossRef CAS Search PubMed
-
(a) A. N. Streletskii, D. G. Permenov, B. B. Bokhonov, A. V. Leonov and S. N. Mudretsova, Colloid J., 2010, 72, 553 CrossRef CAS Search PubMed
- E. Gaffet, F. Faudot and M. Harmelin, Mater. Sci. Forum, 1992, 88–90, 375 CrossRef CAS Search PubMed
- E. Gaffet, Mater. Sci. Eng., A, 1991, 136, 161 CrossRef Search PubMed
- E. Gaffet, F. Faudot and M. Harmelin, Mater. Sci. Eng., A, 1991, 149, 85 CrossRef Search PubMed
- E. Gaffet and J. P. Gaspard, J. Phys., Suppl. Coll. C, 1990, 4, 205 Search PubMed
- E. Gaffet and G. Le Caër, in Encyclopaedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, New York, 2004, vol. 5, pp. 91–129 Search PubMed
- B. J. Pawlak, T. Gregorkiewicz, C. A. J. Ammerlaan, W. Takkenberg, F. D. Tichelaar and P. F. A. Alkemade, Phys. Rev. B: Condens. Matter, 2001, 64, 115308 CrossRef Search PubMed
- T. D. Shen, I. Shmagin, C. C. Koch, R. M. Kolbas, Y. Fahmy, L. Bergman, R. J. Nemanich, M. T. McClure, Z. Sitar and M. X. Quan, Phys. Rev. B: Condens. Matter, 1997, 55, 7615 CrossRef CAS Search PubMed
- A. N. Streletskii, S. N. Mudretsova, A. F. Maiorova, J. C. van Miltenburg and P. Yu. Butyagin, Colloid J., 2001, 63, 635 CrossRef CAS Search PubMed
- A. N. Streletskii, A. V. Leonov, I. V. Beresteskaya, S. S. Mudretsova, A. F. Majorova and P. Yu. Butyagin, Mater. Sci. Forum, 2002, 187, 386 Search PubMed
- C. Diaz-Guerra, A. Montone, J. Piqueras and F. Cardellini, Semicond. Sci. Technol., 2002, 17, 77 CrossRef CAS Search PubMed
- V. Švrček, J.-L. Rehspringer, E. Gaffet, A. Slaoui and J.-C. Muller, J. Cryst. Growth, 2005, 275, 589 CrossRef Search PubMed
- F. Q. Guo and K. Lu, Philos. Mag. Lett., 1998, 77, 181 CrossRef CAS Search PubMed
- G. J. Fan, F. Q. Guo, Z. Q. Hu, M. X. Quan and K. Lu, Phys. Rev. B: Condens. Matter, 1997, 55, 11010 CrossRef CAS Search PubMed
- Y. H. Zhao, Z. H. Jin and K. Lu, Philos. Mag. Lett., 1999, 79, 747 CrossRef CAS Search PubMed
- A. Caro and H. Van Swygenhoven, Phys. Rev. B: Condens. Matter, 2001, 63, 134101 CrossRef Search PubMed
- T. Nasu, F. Araki, O. Uemura, T. Usuki, Y. Kameda, S. Takahashi and K. Tokumitsu, J. Metastable Nanocryst. Mater., 2001, 10, 203 CrossRef Search PubMed
- Y. Tani, Y. Shirakawa, A. Shimosaka and J. Hidaka, J. Non-Cryst. Solids, 2001, 779, 293 Search PubMed
- J. Y. Huang, Acta Mater., 1999, 47, 1801 CrossRef CAS Search PubMed
- N. J. Welham and J. S. Williams, Carbon, 1998, 36, 1309 CrossRef CAS Search PubMed
- T. D. Shen, W. Q. Ge, K. Y. Wang, M. X. Quan, J. T. Wang, W. D. Wei and C. C. Koch, Nanostruct. Mater., 1996, 7, 393 CrossRef CAS Search PubMed
- F. Salver-Disma, J.-M. Tarascon, C. Clinard and J.-N. Rouzaud, Carbon, 1999, 37, 1941 CrossRef CAS Search PubMed
- F. Disma, L. Aymard, L. Dupont and J.-M. Tarascon, J. Electrochem. Soc., 1996, 143, 3959 CrossRef CAS Search PubMed
- F. Salver-Disma, C. Lenain, B. Beaudoin, L. Aymard and J.-M. Tarascon, Solid State Ionics, 1997, 98, 145 CrossRef CAS Search PubMed
- R. Janot and D. Guerard, Carbon, 2002, 40, 2887 CrossRef CAS Search PubMed
-
(a) R. Janot and D. Guerard, Prog. Mater. Sci., 2005, 50, 1 CrossRef CAS Search PubMed
- I. J. Lin and S. Nadiv, Mater. Sci. Eng., 1979, 39, 193 CrossRef CAS Search PubMed
- J. M. Criado and C. Real, Solid State Ionics, 1989, 32–33, 461 CrossRef Search PubMed
- M. Senna, Cryst. Res. Technol., 1985, 20, 209 CrossRef CAS Search PubMed
- A. M. Flank, P. Lagarde, J.-P. Itié, A. Polian and G. R. Hearne, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 224112 CrossRef Search PubMed
- V. Swamy, A. Kuznetsov, L. S. Dubrovinsky, P. F. McMillan, V. B. Prakapenka, G. Shen and B. C. Muddle, Phys. Rev. Lett., 2006, 96, 135702 CrossRef Search PubMed
- T. Isobe and M. Senna, J. Solid State Chem., 1991, 93, 358 CrossRef CAS Search PubMed
- T. Isobe and M. Senna, J. Solid State Chem., 1991, 93, 368 CrossRef CAS Search PubMed
- C. C. Zyryanov, Russ. Chem. Rev., 2008, 77, 105 CrossRef Search PubMed
- V. Šepelák, S. M. Becker, I. Bergmann, S. Indris, M. Scheuermann, A. Feldhoff, C. Kübel, M. Bruns, N. Stürzl, A. S. Ulrich, M. Ghafari, H. Hahn, C. P. Grey, K.-D. Becker and P. Heitjans, J. Mater. Chem., 2012, 22, 3117 RSC
- T. Rojac, B. Malič, M. Kosec, M. Połomska, B. Hilczer, B. Zupančič and B. Zalar, Solid State Ionics, 2012, 215, 1 CrossRef CAS Search PubMed
-
(a) V. Šepelák, D. Baabe, D. Mienert, D. Schultze, F. Krumlich, F. J. Litterst and K. D. Becker, J. Magn. Magn. Mater., 2003, 257, 377 CrossRef Search PubMed
-
(a) M. Menzel, V. Šepelák and K. D. Becker, Solid State Ionics, 2001, 141–142, 663 CrossRef CAS Search PubMed
-
(a) O. Helgason and J. Z. Jiang, Hyperfine Interact., 2002, 139/140, 325 CrossRef CAS Search PubMed
- Z. H. Zhou, J. M. Xue, J. Wang, T. Yu and Z. X. Shen, J. Appl. Phys., 2002, 91, 6015 CrossRef CAS Search PubMed
- C. Jovalekic, M. Zdujic, A. Radakovic and M. Mitric, Mater. Lett., 1995, 24, 365 CrossRef CAS Search PubMed
-
(a) Y. Shi, J. Ding, X. Liu and J. Wang, J. Magn. Magn. Mater., 1999, 205, 249 CrossRef CAS Search PubMed
- J. Ding, X. Y. Liu, J. Wang and Y. Shi, Mater. Lett., 2000, 44, 19 CrossRef CAS Search PubMed
-
(a) Y. Sui, W.-H. Su, F.-L. Zheng and D.-P. Xu, Mater. Sci. Eng., 2000, A286, 115 CAS Search PubMed
- H. M. Widatallah, C. Jonhson, S. H. Al-Harthi, A. M. Gismelseed, A. D. Al-Rawas, S. J. Stewart, M. E. Elzain, I. A. Al-Omari and A. A. Yousif, Hyperfine Interact., 2008, 183, 87 CrossRef CAS Search PubMed
- M. Niyaifar Ramani, M. C. Radhakrishna, A. Hassnpour, M. Mozaffari and J. Amighian, Hyperfine Interact., 2008, 184, 161 CrossRef Search PubMed
- A. González-Angeles, J. Lipka, A. Grusková, V. Jančárik, I. Tóth and J. Sláma, Hyperfine Interact., 2008, 184, 135 CrossRef Search PubMed
- R. A. Borzi, J. Stewart, G. Punte, R. C. Mercader, G. Cernicchiaro and F. Garcia, Hyperfine Interact., 2003, 148/149, 109 CrossRef CAS Search PubMed
- F. J. Berry, X. Ren, J. R. Gancedo and J. F. Marco, Hyperfine Interact., 2004, 156/157, 335 CrossRef CAS Search PubMed
-
(a) S. C. Zanatta, L. F. Cótica, A. Paesano Jr., S. N. de Medeiros, J. B. M. da Cunha and B. Hallouche, J. Am. Ceram. Soc., 2005, 88, 3316 CrossRef CAS Search PubMed
-
(a) L. B. Kong, J. Ma, H. T. Huang, W. Zhu and O. K. Tan, Mater. Lett., 2001, 50, 129 CrossRef CAS Search PubMed
- V. V. Zyryanov, Inorg. Mater., 2000, 36, 54 CrossRef CAS Search PubMed
-
(a) V. V. Zyryanov, Russ. Chem. Rev., 2008, 77, 105 CrossRef CAS Search PubMed
- I. Odeh, A. Fatah, D. Lehlooh and S. H. Mahmood, Hyperfine Interact., 2008, 183, 25 CrossRef CAS Search PubMed
- H. M. Widatallah, A. M. Gismelseed, K. Bouziane, F. J. Berry, A. D. Al Rawas, I. A. AL-Omari, A. A. Yousif and M. E. Elazain, Hyperfine Interact., 2004, 156/157, 223 CrossRef CAS Search PubMed
- G. F. Goya, H. R. Rechenberg and J. Z. Jiang, Mater. Sci. Forum, 1999, 312–314, 545 CrossRef CAS Search PubMed
- L. C. Sánchez, A. M. Calle, J. D. Arboleda, J. J. Beltran, C. A. Barrero, J. Osorio and K. Nomura, Hyperfine Interact., 2008, 183, 117 CrossRef Search PubMed
- M. R. Goldwasser, M. E. Rivas, E. Perez-Zurita, M. L. Cubeiro, A. Grivobal-Constant and G. Leclercq, J. Mol. Catal. A: Chem., 2005, 228, 325 CrossRef CAS Search PubMed
- R. G. Shetkar and A. V. Salker, J. Mater. Sci. Technol., 2010, 26, 1098 CAS Search PubMed
- A. Rajarabar-Darvishi, W.-L. Li, O. Sheikhejad-Bishe, L.-D. Wang, X.-L. Li, N. Li and W.-D. Fei, Trans. Nonferrous Met. Soc. China, 2011, 21, s400 CrossRef Search PubMed
- W. Q. Ni, X. H. Zheng and J. C. Yu, J. Mater. Sci., 2007, 42, 1037 CrossRef CAS Search PubMed
- M. Senna, T. Kinoshita, Y. Abe, H. Kishi, C. Ando, Y. Doshida and B. Stojanovic, J. Eur. Ceram. Soc., 2007, 27, 4301 CrossRef CAS Search PubMed
- B. D. Stojanovic, A. Z. Simoes, C. O. Paiva-Santos, C. Jovalekic, V. V. Mitic and J. A. Varela, J. Eur. Ceram. Soc., 2005, 25, 1985 CrossRef CAS Search PubMed
- K. Wieczorek-Ciurowa and K. Gamrat, Mater. Sci.–Poland, 2007, 25, 219 CAS Search PubMed
- P. Dulian and K. Wieczorek-Ciurowa, Proceedings of 54th PTChem and SITPChem Meeting, Lublin, 2011, p. 347 Search PubMed
- C.-M. Wang, S.-Y. Lin, K.-S. Kao, Y.-C. Chen and S.-C. Weng, J. Alloys Compd., 2010, 491, 423 CrossRef CAS Search PubMed
- W.-X. Yuan and S. K. Hark, J. Eur. Ceram. Soc., 2012, 32, 465 CrossRef CAS Search PubMed
- Y. Yan, L. Jin, L. Feng and G. Cao, Mater. Sci. Eng., B, 2006, 130, 146 CrossRef CAS Search PubMed
- K. Wieczorek-Ciurowa, P. Dulian, A. Nosal and J. Domagała, J. Therm. Anal. Calorim., 2010, 101, 471 CrossRef CAS Search PubMed
- A. F. L. Almeida, R. S. de Oliveira, J. C. Góes, J. M. Sasaki, A. G. Souza Filho, J. Mendes Filho and A. S. B. Sombra, Mater. Sci. Eng., B, 2002, 96, 275 CrossRef Search PubMed
- S. K. Manik and S. K. Pradhan, Phys. E., 2006, 33, 160 CrossRef CAS Search PubMed
- P. Dulian, K. Wieczorek-Ciurowa, W. Bąk and C. Kajtoch, Program and the Book of Abstracts 10th Young Researchers’ Conference Materials Science and Engineering, Belgrade, 2011, p. 12 Search PubMed
- P. Dulian, K. Wieczorek-Ciurowa, W. Bąk, C. Kajtoch and T. Sikora, Book of Abstracts 10th International Conference Solid State Chemistry, Pardubice, 2012, p. 136 Search PubMed
- K. Wieczorek-Ciurowa, P. Dulian, W. Bąk and C. Kajtoch, Przem. Chem., 2011, 90, 1400 CAS Search PubMed
- P. Dulian, K. Wieczorek-Ciurowa, W. Bąk, C. Kajtoch and T. Sikora, Book of Abstracts 10th International Conference Solid State Chemistry, Pardubice, 2012, p. 63 Search PubMed
- E. A. Patterson, S. Kwon, C.-C. Huang and D. P. Cann, Appl. Phys. Lett., 2005, 87, 182911 CrossRef Search PubMed
- S. Kwon, C.-C. Huang, E. A. Patterson and D. P. Cann, Mater. Lett., 2008, 62, 633 CrossRef CAS Search PubMed
- L. Fang, M. Shen, J. Yang and Z. Li, Solid State Commun., 2006, 137, 381 CrossRef CAS Search PubMed
-
(a) J. S. Blakemore, Solid State Physics, Cambridge University Press, Cambridge, UK, 1985 Search PubMed
- L. B. Kong, T. S. Zhang, J. Ma and F. Boey, Prog. Mater. Sci., 2008, 53, 207 CrossRef CAS Search PubMed
- T. Rojac and M. Kosec, in High-Energy Ball Milling: Mechanochemical Processing of Nanopowders, ed. M. Sopicka-Lizer, Woodhead Publishing, Boston, 2010, pp. 113–148 Search PubMed
- K. Uchino, in Ferroelectric Devices, Marcel Dekker, New York, 2000 Search PubMed
- T. R. Shrout and S. J. Zhang, J. Electroceram., 2007, 19, 111 CAS Search PubMed
- J. Rödel, W. Jo, K. T. P. Seifert, E. M. Anton, T. Granzow and D. Damjanovic, J. Am. Ceram. Soc., 2009, 92, 1153 CrossRef Search PubMed
- M. Kosec, B. Malič, A. Benčan and T. Rojac, in Piezoelectric and Acoustic Materials for Transducer Applications, ed. A. Safari and E. K. Akdogan, Springer, New York, 2008, pp. 82–102 Search PubMed
- Y. Saito, H. Takao, T. Tani, T. Nonoyama, K. Takatori, T. Homma, T. Nagaya and M. Nakamura, Nature, 2004, 432, 84 CrossRef CAS Search PubMed
- B. Jaffe, W. R. Cook Jr and H. Jaffe, in Piezoelectric Ceramics, Academic Press, London, New York, 1971 Search PubMed
- E. Hollenstein, D. Damjanovic and N. Setter, J. Eur. Ceram. Soc., 2007, 27, 4093 CrossRef CAS Search PubMed
- K. Kobayashi, Y. Doshida, Y. Mizuno and C. A. Randall, J. Am. Ceram. Soc., 2012, 95, 2928 CrossRef CAS Search PubMed
- U. Flückiger, H. Arend and H. R. Oswald, Am. Ceram. Soc. Bull., 1977, 56, 575 Search PubMed
- K. Kodaira, J. Shimoya, S. Shimada and T. Matsushita, J. Mater. Sci. Lett., 1982, 1, 277 CrossRef CAS Search PubMed
- D. Jenko, A. Benčan, B. Malič, J. Holc and M. Kosec, Microsc. Microanal., 2005, 11, 572 CrossRef CAS Search PubMed
- A. Zhen and J. F. Li, J. Am. Ceram. Soc., 2006, 89, 3669 CrossRef Search PubMed
- T. A. Skidmore and S. J. Milne, J. Mater. Res., 2007, 22, 2265 CrossRef CAS Search PubMed
- N. M. Hagh, B. Jadidian and A. Safari, J. Electroceram., 2007, 18, 339 CrossRef Search PubMed
- Y. Wang, D. Damjanovic, N. Klein and N. Setter, J. Am. Ceram. Soc., 2008, 91, 1962 CrossRef CAS Search PubMed
- A. Reisman, F. Holtzberg and M. Berkenblit, J.
Am. Chem. Soc., 1959, 81, 1292 CrossRef CAS Search PubMed
- Y. Wang, D. Damjanovic, N. Klein, E. Hollenstein and N. Setter, J. Am. Ceram. Soc., 2007, 90, 3485 CrossRef CAS Search PubMed
- V. G. Hill, L. L. Y. Chang and R. I. Harker, J. Am. Ceram. Soc., 1968, 51, 723 CrossRef CAS Search PubMed
- M. Kosec, B. Malič, A. Benčan, T. Rojac and J. Tellier, Funct. Mater. Lett., 2010, 3, 15 CrossRef CAS Search PubMed
- B. Malič, D. Jenko, J. Holc, M. Hrovat and M. Kosec, J. Am. Ceram. Soc., 2008, 91, 1916 CrossRef Search PubMed
- T. F. Soules and R. F. Busbey, J. Chem. Phys., 1981, 75, 969 CrossRef CAS Search PubMed
-
(a) M. Uchida, H. Nakamura, Y. Yamabe-Mitarai and E. Bannai, Scr. Mater., 2005, 52, 11 CrossRef CAS Search PubMed
- D. Jenko, PhD thesis, University of Ljubljana, Slovenia, 2006
- J. M. Sangster and A. D. Pelton, in Critical Coupled Evaluation of Phase Diagrams and Thermodynamic Properties of Binary and Ternary Alkali Salt Systems, American Ceramic Society, Westerville, 1987 Search PubMed
- A. D. Pelton, C. W. Bale and O. L. Lin, Can. J. Chem., 1984, 62, 457 CrossRef CAS Search PubMed
- T. Rojac, A. Benčan, H. Uršič, B. Malič and M. Kosec, J. Am. Ceram. Soc., 2008, 91, 3789 CrossRef CAS Search PubMed
- T. Rojac, A. Benčan and M. Kosec, J. Am. Ceram. Soc., 2010, 93, 1619 CAS Search PubMed
- M. H. Harris and E. K. H. Salje, J. Phys.: Condens. Matter, 1992, 4, 4399 CrossRef CAS Search PubMed
- M. H. Brooker and J. B. Hates, Spectrochim. Acta, 1974, 30A, 2211 CrossRef CAS Search PubMed
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89 CrossRef CAS Search PubMed
- B. M. Gatehouse, S. E. Livingstone and R. S. Nyholm, J. Chem. Soc., 1958, 3137 RSC
- T. Rojac, M. Kosec, P. Šegedin, B. Malič and J. Holc, Solid State Ionics, 2006, 177, 2987 CrossRef CAS Search PubMed
- T. Rojac, M. Kosec, M. Polomska, B. Hilczer, P. Šegedin and A. Benčan, J. Eur. Ceram. Soc., 2009, 29, 2999 CrossRef CAS Search PubMed
- T. Rojac, Ž. Trtnik and M. Kosec, Solid State Ionics, 2011, 190, 1 CrossRef CAS Search PubMed
- T. Rojac, P. Šegedin and M. Kosec, in Infrared Spectroscopy – Materials Science, Engineering and Technology, ed. T. Theophanides, Intech, Rijeka, 2012, pp. 13–42 Search PubMed
- S. Glinšek, B. Malič, T. Rojac, C. Filipič, B. Budič and M. Kosec, J. Am. Ceram. Soc., 2011, 94, 1368 CrossRef Search PubMed
- J. Bernard, A. Benčan, B. Malič, J. Holc, J. Fisher and M. Kosec, in Piezoelectricity for End Users III: Program and Abstracts, Liberec, p. 74 Search PubMed
- N. Klein, E. Hollenstein, D. Damjanovic, H. J. Trodahl and N. Setter, J. Appl. Phys., 2007, 102, 014112 CrossRef Search PubMed
- S. Mrowec, React. Solids, 1988, 5, 241 CrossRef Search PubMed
- P. Baláž, Z. Bastl, T. Havlík, J. Lipka and I. Tóth, Mater. Sci. Forum, 1997, 217, 235 Search PubMed
- P. Baláž, T. Ohtani, Z. Bastl and E. Boldižárová, J. Solid State Chem., 1999, 144, 1 CrossRef Search PubMed
- P. Baláž and E. Godočíková, J. Therm. Anal. Calorim., 2001, 65, 51 CrossRef Search PubMed
- P. Baláž, J. Z. Jiang, L. Takacs, E. Godočíková, E. Boldižárová, Z. Bastl and M. Luxová, Czech. J. Phys., 2002, 52, A65 CrossRef Search PubMed
- P. Baláž, L. Takacs, J. Z. Jiang, V. Soika and M. Luxová, J. Metastable Nanocryst. Mater., 2002, 13, 257 CrossRef Search PubMed
- P. Baláž, L. Takacs, T. Ohtani, D. E. Mack, E. Boldižárová, V. Soika and M. Achimovičová, J. Alloys Compd., 2002, 337, 76 CrossRef Search PubMed
- W. Wang, Y. Liu, Y. Zhan, Ch. Zheng and G. Wang, Mater. Res. Bull., 2001, 36, 1977 CrossRef CAS Search PubMed
- D. Wang, D. Yu, M. Mo, X. Liu and Y. Qian, Solid State Commun., 2003, 125, 475 CrossRef CAS Search PubMed
- J. Xiang, S. H. Yu, B. Liu, Y. Xu, X. Gen and L. Ren, Inorg. Chem. Commun., 2004, 7, 572 CrossRef CAS Search PubMed
- Q. Liu, Y. Ni, G. Yin, J. Hong and Z. Xu, Mater. Chem. Phys., 2005, 89, 379 CrossRef CAS Search PubMed
- S. A. Chen and W. M. Liu, Langmuir, 1999, 15, 8100 CrossRef CAS Search PubMed
- N. A. Dhas, A. Zaban and A. Gedanken, Chem. Mater., 1999, 11, 806 CrossRef CAS Search PubMed
- Z. P. Qiao, Y. Xie, Y. T. Qian and Y. Zhu, Mater. Chem. Phys., 2000, 62, 88 CrossRef CAS Search PubMed
- D. B. Zhang, L. M. Qi, H. M. Cheng and J. Ma, J. Colloid Interface Sci., 2002, 246, 413 CrossRef CAS Search PubMed
- N. R. Pawaskar, S. D. Sathaye, M. Bhadbhade and K. R. Patil, Mater. Res. Bull., 2002, 37, 1539 CrossRef CAS Search PubMed
- Ch. Lan, K. Hong, W. Wang and G. Wang, Solid State Commun., 2003, 125, 455 CrossRef CAS Search PubMed
- J. Chen, Y. Li, Y. Wang, J. Yun and D. Cao, Mater. Res. Bull., 2004, 39, 185 CrossRef CAS Search PubMed
- A. M. Arriaga, O. Fernandez and O. Solorza, Int. J. Hydrogen Energy, 1998, 23, 995 CrossRef Search PubMed
- R. He, X. Qian, J. Yin, H. Xi, L. Bian and Z. Zhu, Colloids Surf., A, 2003, 220, 151 CrossRef CAS Search PubMed
- L. Foglia, L. Suber and M. Righini, Colloids Surf., A, 2001, 177, 3 CrossRef Search PubMed
- L. Hao, M. You, X. Mo, W. Jiang, Y. Zhu, Y. Zhou, Y. Hu, X. Liu and Z. Chen, Mater. Res. Bull., 2003, 38, 723 CrossRef CAS Search PubMed
- Ch. Barglik-Chory, D. Buchold, M. Schmitt, W. Kiefer, C. Heske, C. Kumpf, O. Fuchs, L. Weinhardt, A. Stahl, E. Umbach, M. Lentze, J. Geurts and G. Müller, Chem. Phys. Lett., 2003, 379, 443 CrossRef CAS Search PubMed
- W. Wang, Z. Liu, Ch. Zheng, C. Xu, Y. Liu and G. Wang, Mater. Lett., 2003, 57, 2755 CrossRef CAS Search PubMed
- B. Yan, D. Chen and X. Jiao, Mater. Res. Bull., 2004, 39, 1655 CrossRef CAS Search PubMed
- A. E. Raevskaya, A. L. Stroyuk, S. Ya, Kuchmii and A. I. Kryukov, J. Mol. Catal. A: Chem., 2004, 212, 259 CrossRef CAS Search PubMed
- L. Gao, E. Wang, S. Lian, Z. Kang, Y. Lan and D. Wu, Solid State Commun., 2004, 130, 309 CrossRef CAS Search PubMed
- Ch. Xu, Z. Zhang and Q. Ye, Mater. Lett., 2004, 58, 1671 CrossRef CAS Search PubMed
- S. Reimann and M. Manninen, Rev. Mod. Phys., 2004, 74, 1283 CrossRef Search PubMed
- A. P. Alivisatos, Science, 1996, 271, 933 CAS Search PubMed
- P. Baláž, T. Havlík, Z. Bastl and J. Briančin, J. Mater. Sci. Lett., 1995, 14, 344 CrossRef Search PubMed
- P. Baláž, T. Havlík, J. Briančin and R. Kammel, Scr. Metall. Mater., 1995, 32, 1357 CrossRef Search PubMed
- P. Baláž, T. Havlík, Z. Bastl, J. Briančin and R. Kammel, J. Mater. Sci. Lett., 1996, 15, 1161 CrossRef Search PubMed
- P. Baláž, M. Bálintová, Z. Bastl, J. Briančin and V. Šepelák, Solid State Ionics, 1997, 101–103, 45 CrossRef Search PubMed
- P. Baláž and T. Ohtani, Mater. Sci. Forum, 2000, 343–346, 389 CrossRef Search PubMed
- P. Baláž, E. Boldižárová, E. Godočíková and J. Briančin, Mater. Lett., 2003, 57, 1585 CrossRef Search PubMed
- E. Godočíková, P. Baláž and E. Gock, Acta Metall. Slovaca, 2004, 10, 73 Search PubMed
- P. Baláž, E. Boldižárová and E. Godočíková, Mater. Sci. Forum, 2005, 480–481, 453 CrossRef Search PubMed
- P. Baláž, E. Godočíková, L. Kril'ová, P. Lobotka and E. Gock, Mater. Sci. Eng., A, 2004, 386, 442 Search PubMed
- E. Godočíková, P. Baláž, J. M. Criado, C. Real and E. Gock, Thermochim. Acta, 2006, 440, 19 CrossRef Search PubMed
- E. Godočíková, P. Baláž, E. Gock, W. S. Choi and B. S. Kim, Powder Technol., 2006, 164, 147 CrossRef Search PubMed
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttall, J. Phys. Chem. Ref. Data, 1982, 11(suppl. 2), 1–392 Search PubMed
- E. Dutková, P. Baláž, P. Pourghahramani, S. Velumani, J. A. Ascencio and N. G. Kostova, J. Nanosci. Nanotechnol., 2009, 9, 6600–6605 CrossRef Search PubMed
- B. E. Warren and B. L. Averbach, J. Appl. Phys., 1950, 21, 595 CrossRef CAS Search PubMed
- M. José-Yacamán, J. A. Ascencio and H. Lin, J. Vac. Sci. Technol., 2001, B19, 1091 Search PubMed
- J. A. Ascencio, C. Gutierrez-Wing, M. E. Espinosa-Pesqueira, M. Marin, S. Tehuacanero, C. Zorilla and M. José-Yacamán, Surf. Sci., 1998, 396, 349 CrossRef CAS Search PubMed
- J. A. Ascencio, H. B. Lin, U. Pal, A. Medina and Z. L. Wang, Microsc. Res. Tech., 2006, 69, 522 CrossRef CAS Search PubMed
- E. Dutková, P. Baláž and P. Pourghahramani, J. Optoelectron. Adv. Mater., 2009, 11(12), 2102 Search PubMed
- E. Dutková, P. Baláž, P. Pourghahramani, A. V. Nguyen, V. Šepelák, A. Feldhoff, J. Kováč and A. Šatka, Solid State Ionics, 2008, 179, 1242 CrossRef Search PubMed
- E. Dutková, P. Baláž, P. Pourghahramani, V. Balek, A. V. Nguyen, A. Šatka, J. Kováč and J. Ficeriová, J. Nano Res., 2012, 18–19, 247 Search PubMed
- P. Baláž, P. Pourghahramani, E. Dutková, M. Fabián, J. Kováč and A. Šatka, Cent. Eur. J. Chem., 2009, 7(2), 215 CrossRef Search PubMed
- P. Baláž, P. Pourghahramani, M. Achimovičová, E. Dutková, J. Kováč, A. Šatka and J. Z. Jiang, J. Cryst. Growth, 2011, 332, 1 CrossRef Search PubMed
- E. Dutková, L. Takacs, M. J. Sayagues, P. Baláž, J. Kováč and A. Šatka, Chem. Eng. Sci., 2013, 85, 25 CrossRef Search PubMed
- O. Kubaschewski and L. L. Evans, Metallurgical Thermochemistry, Pergamon Press, London, 1955 Search PubMed
- C. Wang, W. X. Zhang, X. F. Qian, X. M. Zhang, Y. Xie and Y. T. Qian, Mater. Chem. Phys., 1999, 60, 99 CrossRef CAS Search PubMed
- M. Achimovičová, P. Baláž, J. Ďurišin, N. Daneu, J. Kováč, A. Šatka, A. Feldhoff and E. Gock, Int. J. Mater. Res., 2011, 102, 441 CrossRef Search PubMed
- K. Chung, D. Wamwangi, M. Woda, M. Wuttig and W. Bensch, J. Appl. Phys., 2008, 103, 083523 CrossRef Search PubMed
- B. Pejova, I. Grozdanov and A. Tanuševski, Mater. Chem. Phys., 2004, 83, 245 CrossRef CAS Search PubMed
- S. K. Mishra, S. Satpathy and O. Jepsen, J. Phys.: Condens. Matter, 1997, 9, 461 CrossRef CAS Search PubMed
- M. Achimovičová, P. Baláž, T. Ohtani, N. Kostova, G. Tyuliev, A. Feldhoff and V. Šepelák, Solid State Ionics, 2011, 192, 632 CrossRef Search PubMed
- M. Achimovičová, N. Daneu, A. Rečnik, J. Ďurišin, P. Baláž, M. Fabián, J. Kováč and A. Šatka, Chem. Pap., 2009, 63, 562 CrossRef Search PubMed
- M. Achimovičová, A. Rečnik, M. Fabián and P. Baláž, Acta Montan. Slovaca, 2011, 16, 147 Search PubMed
- H. Wang and F. Du, Cryst. Res. Technol., 2006, 41, 323 CrossRef CAS Search PubMed
- A. Sashchiuk, L. Amirav, M. Bashouti, M. Krueger, U. Sivan and E. Lifshitz, Nano Lett., 2004, 4, 159 CrossRef CAS Search PubMed
- M. Achimovičová, P. Baláž, E. Dutková, E. Gock and V. Šepelák, J. Balkan Tribol. Assoc., 2009, 15, 79 Search PubMed
- M. Achimovičová, K. L. da Silva, N. Daneu, A. Rečnik, S. Indris, H. Hain, M. Scheuermann, H. Hahn and V. Šepelák, J. Mater. Chem., 2011, 21, 5873 RSC
- P. Hu, Y. Cao, D. Jia and L. Wang, Mater. Lett., 2010, 64, 493 CrossRef CAS Search PubMed
- M. Achimovičová, F. J. Gotor, C. Real and N. Daneu, J. Mater. Sci.: Mater. Electron., 2012, 23, 1844 CrossRef Search PubMed
- V. M. Garcia, M. T. S. Nair, P. K. Nair and R. A. Zingaro, Semicond. Sci. Technol., 1997, 12, 645 CrossRef CAS Search PubMed
- P. Matteazzi and G. Le Caër, J. Am. Ceram. Soc., 1991, 74, 1382 CrossRef CAS Search PubMed
- Z. G. Liu, L. L. Ye, J. T. Guo, G. S. Li and Z. Q. Hu, J. Mater. Res., 1995, 10, 3129 CrossRef CAS Search PubMed
- M. S. El-Eskandarany, J. Alloys Compd., 2000, 305, 225 CrossRef CAS Search PubMed
- X. Cui, Y. Wang, L. Wang, L. Cui and M. Qi, Pet. Sci. Technol., 2001, 19, 971 CrossRef CAS Search PubMed
- B. H. Lohse, A. Calka and D. Wexler, J. Alloys Compd., 2005, 394, 148 CrossRef CAS Search PubMed
- B. Li, L. Cui, Y. Zheng and Ch. Xu, Chin. J. Chem. Eng., 2007, 15, 138 CrossRef CAS Search PubMed
- H. Jia, Z. Zhang, Z. Qia, G. Liu and X. Bian, J. Alloys Compd., 2009, 472, 97 CrossRef CAS Search PubMed
- Q. Yuan, Y. Zheng and H. Yu, Int. J. Refract. Met. Hard Mater., 2009, 27, 696 CrossRef CAS Search PubMed
- B. Ghosh and S. K. Pradhan, Mater. Chem. Phys., 2010, 120, 537 CrossRef CAS Search PubMed
- M. B. Rahaei, R. Y. Rad, A. Kazemzadeh and T. Ebadzadeh, Powder Technol., 2012, 217, 369 CrossRef CAS Search PubMed
- T. S. Suzuki and M. Nagumo, Scr. Metall. Mater., 1995, 32, 1215 CrossRef CAS Search PubMed
- J. Y. Xiang, S. C. Liu, W. T. Hu, Y. Zhang, C. K. Chen, P. Wang, J. L. He, D. L. Yu, B. Xu, Y. F. Lu, Y. J. Tian and Z. Y. Liu, J. Eur. Ceram. Soc., 2011, 31, 1491 CrossRef CAS Search PubMed
-
(a) B. K. Yen, J. Alloys Compd., 1998, 268, 266 CrossRef CAS Search PubMed
- A. A. Mahday, M. S. El-Eskandarany, H. A. Ahmed and A. A. Amer, J. Alloys Compd., 2000, 299, 244 CrossRef CAS Search PubMed
- L. Takacs, J. Solid State Chem., 1996, 125, 75 CrossRef CAS Search PubMed
- A. Calka and W. A. Kaczmarek, Scr. Metall. Mater., 1992, 26, 249 CrossRef CAS Search PubMed
- B. R. Murphy and T. H. Courtney, J. Mater. Res., 1999, 14, 4274 CrossRef CAS Search PubMed
- L. Liu and Y. D. Dong, Nanostruct. Mater., 1993, 2, 463 CrossRef CAS Search PubMed
- S. I. Cha and S. H. Hong, J. Metastable Nanocryst. Mater., 2003, 15–16, 319 CrossRef CAS Search PubMed
- S. Bolokang, C. Banganayi and M. Phasha, Int. J. Refract. Met. Hard Mater., 2010, 28, 211 CrossRef CAS Search PubMed
- S. Gomari and S. Sharafi, J. Alloys Compd., 2010, 490, 26 CrossRef CAS Search PubMed
- M. S. El-Eskandarany, K. Sumiyama and K. Suzuki, J. Mater. Res., 1995, 10, 659 CrossRef Search PubMed
- Z. G. Yang and L. L. Sbaw, Nanostruct. Mater., 1996, 7, 813 CrossRef Search PubMed
- H. Abderrazak and M. Abdellaoui, Mater. Lett., 2008, 62, 3839 CrossRef CAS Search PubMed
- B. Ghosh and S. K. Pradhan, J. Alloys Compd., 2009, 486, 480 CrossRef CAS Search PubMed
- G. Zhang, G. Wei, K. Zheng, L. Li, D. Xu, D. Wang, Y. Xue and W. Su, J. Nanosci. Nanotechnol., 2010, 10, 1951 CrossRef CAS Search PubMed
- K. Kudaka, K. Iizumi and T. Sasaki, J. Ceram. Soc. Jpn., 1999, 107, 1019 CrossRef CAS Search PubMed
- A. Calka, Appl. Phys. Lett., 1991, 59, 1568 CrossRef CAS Search PubMed
- Y. Ogino, T. Yamasaki, N. Atzumi and K. Yoshioka, Mater. Trans., JIM, 1993, 35, 1212 Search PubMed
- Z. H. Chin and T. P. Perng, Appl. Phys. Lett., 1997, 70, 2380 CrossRef CAS Search PubMed
- F. J. Gotor, M. D. Alcala, C. Real and J. M. Criado, J. Mater. Res., 2002, 17, 1655 CrossRef CAS Search PubMed
- M. S. El-Eskandarany and A. H. Ashour, J. Alloys Compd., 2000, 313, 224 CrossRef CAS Search PubMed
- M. A. Bab, L. Mendoza-Zelis and L. C. Damonte, Acta Mater., 2001, 49, 4205 CrossRef CAS Search PubMed
- A. Calka and J. S. Williams, Mater. Sci. Forum, 1992, 88–90, 787 CrossRef CAS Search PubMed
- M. A. Roldan, V. Lopez-Flores, M. D. Alcala, A. Ortega and C. Real, J. Eur. Ceram. Soc., 2010, 30, 2099 CrossRef CAS Search PubMed
- L. Liu, L. Lu, L. Chen, Y. Qin and L. D. Zhang, Metall. Mater. Trans. A, 1999, 30, 1097 CrossRef Search PubMed
- M. S. El-Eskandarany, K. Sumiyama, K. Aoki, T. Masumoto and K. Suzuki, J. Mater. Res., 1994, 9, 2891 CrossRef CAS Search PubMed
- I. Lucks, P. Lamparter and E. J. Mittemeijer, Acta Mater., 2001, 49, 2419 CrossRef CAS Search PubMed
- C. Real, M. A. Roldan, M. D. Alcala and A. Ortega, J. Am. Ceram. Soc., 2007, 90, 3085 CrossRef CAS Search PubMed
- H. Zhang, E. H. Kisi and S. Myhra, J. Phys. D: Appl. Phys., 1996, 29, 1367 CrossRef CAS Search PubMed
- H. Yang and P. G. McCormick, J. Mater. Sci., 1993, 28, 5663 CrossRef CAS Search PubMed
- C. J. Lu, J. Zhang and Z. Q. Li, J. Alloys Compd., 2004, 381, 278 CrossRef CAS Search PubMed
- Y. Chen, Z. L. Li and J. S. Williams, J. Mater. Sci. Lett., 1995, 14, 542 CrossRef CAS Search PubMed
- S. J. Campbell, M. Hofmann and A. Calka, Phys. B, 2000, 276–278, 899 CrossRef CAS Search PubMed
- Y. Chen and J. S. Williams, J. Mater. Res., 1996, 11, 1500 CrossRef CAS Search PubMed
- A. Calka, S. W. Wilkins, H. Hashizume, D. J. Cookson and J. I. Nikolov, Mater. Sci. Forum, 1997, 235–238, 517 CrossRef CAS Search PubMed
- A. Calka and J. I. Nikolov, Nanostruct. Mater., 1995, 6, 409 CrossRef CAS Search PubMed
- J. F. Sun, M. Z. Wang, Y. C. Zhao, X. P. Li and B. Y. Liang, J. Alloys Compd., 2009, 482, L29 CrossRef CAS Search PubMed
- W. Zhang, Z. Li and D. Zhang, J. Mater. Res., 2010, 25, 464 CrossRef CAS Search PubMed
- A. Calka and A. P. Radlinski, J. Less Common Met., 1990, 161, L23 CrossRef CAS Search PubMed
- L. L. Ye, Z. G. Liu, M. X. Quan and Z. Q. Hu, J. Appl. Phys., 1996, 80, 1910 CrossRef CAS Search PubMed
- M. A. Aviles, J. M. Cordoba, M. J. Sayagues and F. J. Gotor, Ceram. Int., 2011, 37, 1895 CrossRef CAS Search PubMed
- S. Licht, C. Hettige, J. Lau, U. Cubeta, H. Wu, J. Stuart and B. Wanga, Electrochem. Solid-State Lett., 2012, 15, A12 CrossRef CAS Search PubMed
- M. A. Morris and D. G. Morris, J. Mater. Sci., 1991, 26, 4687 CrossRef CAS Search PubMed
- K. Iizumi, C. Sekiya, S. Okada, K. Kudou and T. Shishido, J. Eur. Ceram. Soc., 2006, 26, 635 CrossRef CAS Search PubMed
- K. Kudaka, K. Iizumi, T. Sasaki and S. Okada, J. Alloys Compd., 2001, 315, 104 CrossRef CAS Search PubMed
- D. D. Radev and D. Klissurski, J. Alloys Compd., 1994, 206, 39 CrossRef CAS Search PubMed
- Z. H. Yan, M. Oehring and R. Bormann, J. Appl. Phys., 1992, 72, 2478 CrossRef CAS Search PubMed
- B. K. Yen and T. Aizawa, J. Am. Ceram. Soc., 1998, 81, 1953 CrossRef CAS Search PubMed
- S. Doppiu, M. Monagheddu, G. Cocco, F. Maglia, U. Anselmi-Tamburini and Z. A. Munir, J. Mater. Res., 2001, 16, 1266 CrossRef CAS Search PubMed
- B. K. Yen, T. Aizawa and J. Kihara, Mater. Sci. Eng., A, 1996, 220, 8 CrossRef Search PubMed
- L. Liu, S. Casadio, M. Magín and C. A. Nannetti, J. Alloys Compd., 1995, 227, 76 CrossRef CAS Search PubMed
- B. K. Yen, T. Aizawa, J. Kihara and N. Sakakibara, Mater. Sci. Eng., A, 1997, 239–240, 515 CrossRef Search PubMed
- T. Lou, G. Fan, B. Ding and Z. Hu, J. Mater. Res., 1997, 12, 1172 CrossRef CAS Search PubMed
- E. Ma, J. Pagan, G. Cranford and M. Atzmon, J. Mater. Res., 1993, 8, 1836 CrossRef CAS Search PubMed
- B. B. Bokhonov, I. G. Konstanchuk and V. V. Boldyrev, J. Alloys Compd., 1995, 218, 190 CrossRef CAS Search PubMed
- L. Liu, F. Padella, W. Guo and M. Magini, Acta Metall. Mater., 1995, 43, 3755 CrossRef CAS Search PubMed
- P. C. Kang and Z. D. Yin, Nanotechnology, 2004, 15, 851 CrossRef CAS Search PubMed
- G. T. Fei, L. Liu, X. Z. Ding, L. D. Zhang and Q. Q. Zheng, J. Alloys Compd., 1995, 229, 280 CrossRef CAS Search PubMed
- H. G. Tang, X. F. Ma, W. Zhao, X. W. Yan and J. M. Yan, Mater. Res. Bull., 2004, 39, 707 CrossRef CAS Search PubMed
- M. A. Roldan, M. D. Alcala and C. Real, Ceram. Int., 2012, 38, 687 CrossRef CAS Search PubMed
- M. Miki, T. Yamasaki and Y. Ogino, Mater. Trans., JIM, 1993, 34, 952 CAS Search PubMed
- M. Miki, T. Yamasaki and Y. Ogino, Mater. Trans., JIM, 1992, 33, 839 CAS Search PubMed
- J. M. Cordoba, M. J. Sayagues, M. D. Alcala and F. J. Gotor, J. Am. Ceram. Soc., 2005, 88, 1760 CrossRef CAS Search PubMed
- J. M. Cordoba, M. J. Sayagues, M. D. Alcala and F. J. Gotor, J. Am. Ceram. Soc., 2007, 90, 381 CrossRef CAS Search PubMed
- J. M. Cordoba, M. A. Aviles, M. J. Sayagues, M. D. Alcala and F. J. Gotor, J. Alloys Compd., 2009, 482, 349 CrossRef CAS Search PubMed
- J. M. Cordoba, M. J. Sayagues, M. D. Alcala and F. J. Gotor, J. Mater. Chem., 2007, 17, 650 RSC
- C. J. Lua and Z. Q. Li, J. Alloys Compd., 2008, 461, 216 CrossRef Search PubMed
- M. A. Aviles, J. M. Cordoba, M. J. Sayagues, M. D. Alcala and F. J. Gotor, J. Am. Ceram. Soc., 2010, 93, 696 CrossRef CAS Search PubMed
- A. Mosbah, A. Calka and D. Wexler, J. Alloys Compd., 2006, 424, 279 CrossRef CAS Search PubMed
- C. Deidda, F. Delogu and G. Cocco, J. Metastable Nanocryst. Mater., 2004, 20–21, 337 CrossRef CAS Search PubMed
- E. M. Sharifi, F. Karimzadeh and M. H. Enayati, Adv. Powder Technol., 2011, 22, 354 CrossRef CAS Search PubMed
- B. Zhang and Z. Q. Li, J. Alloys Compd., 2005, 392, 183 CrossRef CAS Search PubMed
- A. A. Popovich, V. P. Reva and V. N. Vasilenko, J. Alloys Compd., 1993, 190, 143 CrossRef CAS Search PubMed
- M. S. El-Eskandarany, J. Alloys Compd., 2000, 296, 175 CrossRef CAS Search PubMed
- N. J. Welham, J. Am. Ceram. Soc., 2000, 83, 1290 CrossRef CAS Search PubMed
- R. Ricceri and P. Matteazzi, Mater. Sci. Eng., A, 2004, 379, 341 CrossRef Search PubMed
- E. Bilgi, H. E. Camurlu, B. Akgun, Y. Topkaya and N. Sevinc, Mater. Res. Bull., 2008, 43, 873 CrossRef CAS Search PubMed
- N. Setoudeh and N. J. Welham, J. Alloys Compd., 2006, 420, 225 CrossRef CAS Search PubMed
- B. Akgun, H. E. Camurlu, Y. Topkaya and N. Sevinc, Int. J. Refract. Met. Hard Mater., 2011, 29, 601 CrossRef Search PubMed
- Y. Sun, B. Yao, Q. He, F. Su and H. Z. Wang, J. Alloys Compd., 2009, 479, 599 CrossRef CAS Search PubMed
- J. W. Kim, J. H. Shim, J. P. Ahn, Y. W. Cho, J. H. Kim and K. H. Oh, Mater. Lett., 2008, 62, 2461 CrossRef CAS Search PubMed
- J. David Newell and S. N. Patankar, Mater. Lett., 2009, 63, 81 CrossRef Search PubMed
- T. Dasgupta and A. M. Umarji, J. Alloys Compd., 2008, 461, 292 CrossRef CAS Search PubMed
- T. F. Grigorieva, A. P. Barinova and N. Z. Lyakhov, J. Nanopart. Res., 2003, 5, 439 CrossRef CAS Search PubMed
-
(a) A. N. Streletskii, I. V. Kolbanev, A. B. Borunova and P. Yu. Butyagin, J. Mater. Sci., 2004, 39, 5175 CrossRef CAS Search PubMed
-
(a) B. Bokhonov and M. Korchagin, J. Alloys Compd., 2002, 333, 308 CrossRef CAS Search PubMed
- A. N. Streletskii, O. S. Morozova, I. V. Berestetskaya and A. B. Borunova, Mater. Sci. Forum, 1998, 269–272, 283 CrossRef CAS Search PubMed
- Ch. Borchers, A. V. Leonov, T. I. Khomenko and O. S. Morozova, J. Phys. Chem. B, 2005, 109, 10341 CrossRef CAS Search PubMed
- C. Borchers, T. I. Khomenko, O. S. Morozova, A. V. Galakhov, E. Z. Kurmaev, J. McNaughton, M. V. Yabloskikh and A. Moewes, J. Phys. Chem. B, 2006, 110, 196 CrossRef CAS Search PubMed
-
(a) H. Imamura, M. Kusuhara, S. Minami, M. Matsumoko, K. Masanari, Y. Sakata, K. Itoh and T. Fukunada, Acta Mater., 2003, 51, 6407 CrossRef CAS Search PubMed
- B. Bokhonov, M. Korchagin and Yu. Borisova, J. Alloys Compd., 2004, 372, 141 CrossRef CAS Search PubMed
-
(a) A. B. Borunova, A. N. Streletskii, S. N. Mudretsova, A. V. Leonov and P. Yu. Butyagin, Colloid J., 2011, 73, 605 CrossRef CAS Search PubMed
- I. V. Povstugar, A. N. Streletskii, D. G. Permenov, I. V. Kolbanev and S. N. Mudretsova, J. Alloys Compd., 2009, 483, 298 CrossRef CAS Search PubMed
- Ch. Borchers, O. S. Morozova, T. I. Khomenko, A. V. Leonov, A. V. Postnikov, E. Z. Kurmaev, A. Moewes and A. Pundt, J. Phys. Chem. C, 2008, 112, 5869 CAS Search PubMed
- B. Bokhonov, Yu. Borisova and M. Korchagin, Carbon, 2004, 42, 2067 CrossRef CAS Search PubMed
- A. N. Streletskii, I. V. Kolbanev, A. B. Borunova, A. V. Leonov and P. Yu. Butjagin, Colloid J., 2004, 66, 729 CrossRef CAS Search PubMed
- A. Pivkina, A. N. Streletskii, I. Kolbanev, P. Ul’yanova, Yu. Frolov, P. Yu. Butyagin and J. Schoonman, J. Mater. Sci., 2004, 39, 5451 CrossRef CAS Search PubMed
- A. N. Streletskii, I. V. Kolbanev, A. B. Borunova and P. Yu. Butyagin, Colloid J., 2005, 67, 631 CrossRef CAS Search PubMed
-
(a) X. N. Huang, C. J. Lu, Y. Wang, H. Y. Shen, D. Chen and Y. X. Huang, Int. J. Hydrogen Energy, 2012, 37, 7457 CrossRef CAS Search PubMed
- E. I. Shkolnikov, A. Z. Zhuk and M. S. Vlaskin, Renewable Sustainable Energy Rev., 2011, 15, 4611 CrossRef CAS Search PubMed
- A. N. Streletskii, S. N. Mudretsova, I. V. Povstugar and P. Yu. Butyagin, Colloid J., 2006, 68, 623 CrossRef CAS Search PubMed
- A. Yu. Dolgoborodov, M. F. Gogulya, M. N. Makhov, I. V. Kolbanev and A. N. Streletskii, Proceedings of 29th International Pyrotechnics Seminar, IP-SUSA, Inc., 2002, p. 557 Search PubMed
-
(a) A. Y. u. Dolgoborodov, M. N. Makhov, A. N. Streletskii, I. V. Kolbanev and M. F. Gogulya, Russian Patent, 2235085, 2004 Search PubMed
-
(a) E. L. Dreizin, Prog. Energy Combust. Sci., 2009, 35, 141 CrossRef CAS Search PubMed
-
(a) Y. Wang, W. Jiang, X. Zhang, H. Liu, Y. Liu and F. Li, Thermochim. Acta, 2011, 512, 233 CrossRef CAS Search PubMed
- A. N. Streletskii, I. V. Kolbanev and P. Y. u. Butyagin, Book of abstracts of 19th International Symposium on Metastable, Amorphous and Nanostructured Materials ISMANAM, Moscow, 2012, p. 41 Search PubMed
- A. Caro and H. Van Swygenhoven, Phys. Rev. B: Condens. Matter, 2001, 63, 134101 CrossRef Search PubMed
-
(a) T. H. Courtney, Mater. Trans., JIM, 1995, 36, 110 CAS Search PubMed
- C. Suryanarayana, Rev. Adv. Mater. Sci., 2008, 18, 203 CAS Search PubMed
- J. L. H. Rivera, J. J. C. Rivera, V. P. del Angel, V. G. Febles, O. C. Alonso and R. Martínez-Sánchez, Mater. Des., 2012, 37, 96 CrossRef Search PubMed
- C. Suryanarayana, J. Alloys Compd., 2011, 509, S229 CrossRef CAS Search PubMed
- M. F. Zawrah, H. Abdel-Kader and N. E. Elbaly, Mater. Res. Bull., 2012, 47, 655 CrossRef CAS Search PubMed
- A. M. Soufiani, M. H. Enayati and F. Karimzadeh, Mater. Des., 2010, 31, 3954 CrossRef Search PubMed
- M. Klimenkov, J. Nucl. Mater., 2010, 217 Search PubMed
- A. H. Monazzah, A. Simchi and S. M. S. Reihani, Mater. Sci. Eng., A, 2010, 527, 2567 CrossRef Search PubMed
- S. Alamolhoda, S. Heshmati-Manesh and A. Ataie, Adv. Powder Technol., 2012, 23, 343 CrossRef CAS Search PubMed
- M. Z. Mehrizi, A. Saidi and M. Shamanian, Mater. Sci. Technol., 2011, 27, 1465 CrossRef CAS Search PubMed
- D. D. Gu, W. Meiners, C. Li and Y. F. Shen, Mater. Sci. Eng., A, 2010, 527, 6340 CrossRef Search PubMed
- D. D. Gu, W. Meiners, Y. C. Hagedorn, K. Wissenbach and R. Poprawe, J. Phys. D: Appl. Phys., 2010, 43, 135402 CrossRef Search PubMed
- L. H. Tian, C. X. Li, C. J. Li and G. J. Yang, J. Therm. Spray Technol., 2012, 21, 689 CrossRef CAS Search PubMed
- M. Sheikhzadeh and S. Sanjabi, Mater. Des., 2012, 39, 366 CrossRef CAS Search PubMed
- D. D. Gu, Z. Y. Wang, Y. F. Shen, Q. Li and Y. F. Li, Appl. Surf. Sci., 2009, 255, 9230 CrossRef CAS Search PubMed
- A. Miklaaszewski, M. U. Jurczyk and M. Jurczyk, Solid State Phenom., 2012, 183, 131 CrossRef Search PubMed
- E. H. Jazi, G. Borhani and R. E. Farsani, Micro Nano Lett., 2012, 7, 448 Search PubMed
- X. T. Luo, G. J. Yang and C. J. Li, Powder Technol., 2011, 217, 591 CrossRef Search PubMed
- Y. F. Wu, G. Y. Kim and A. M. Russell, Mater. Sci. Eng., A, 2012, 532, 558 CrossRef CAS Search PubMed
- N. Banno and T. Takeuchi, J. Jpn. Inst. Met., 2009, 73, 651 CrossRef CAS Search PubMed
- F. C. R. Hernández and H. A. Calderón, Mater. Chem. Phys., 2012, 132, 815 CrossRef Search PubMed
- N. Sekido, A. Hoshino, M. Fukuzaki, Y. Yamabe-Mitari and T. Maruko, Mater. Sci. Eng., A, 2011, 528, 8451 CrossRef CAS Search PubMed
- E. M. Salleh and Z. Hussain, Key Eng. Mater., 2011, 471–472, 798 CrossRef CAS Search PubMed
- H. Zuhailawati, H. M. Salihin and Y. Mahani, J. Alloys Compd., 2010, 489, 369 CrossRef CAS Search PubMed
- C. Li, D. D. Gu, Y. F. Shen, G. B. Meng and Y. F. Li, Adv. Eng. Mater., 2011, 13, 418 CrossRef CAS Search PubMed
- F. Shojaeepour, P. Abachi, K. Purazrang and A. H. Moghanian, Powder Technol., 2012, 222, 80 CrossRef CAS Search PubMed
- P. Sahani, S. Mula, P. K. Roi, P. C. Kang and C. C. Koch, Mater. Sci. Eng., A, 2011, 528, 7781 CrossRef CAS Search PubMed
- M. Razavi, M. R. Rahimipour and R. Yazdani-Rad, Adv. Appl. Ceram., 2011, 110, 367 CrossRef CAS Search PubMed
- Y. Y. Wu, X. F. Liu, G. L. Ma, C. Li and J. Q. Zhang, J. Alloys Compd., 2010, 497, 139 CrossRef CAS Search PubMed
- Y. D. Kim, S.-T. Oh, K. H. Min, H. Jeon and I.-H. Moon, Scr. Mater., 2001, 44, 293 CrossRef CAS Search PubMed
- J. W. Lee, Z. A. Munir and M. Ohyanagi, Mater. Sci. Eng., A, 2002, 325, 221 CrossRef Search PubMed
- M. El-Eskandarany, M. Omori, T. J. Konno, K. Sumiyama, T. Hirai and K. Suzuki, Metall. Mater. Trans. A, 2001, 32, 157 CrossRef Search PubMed
- S. D. de la Torre, D. E. Garcia, N. Claussen, R. Janssen, Y. Nishikawa, H. Miyamoto, R. Martinez-Sanchez, A. Garcia-Luna and D. Rios-Jara, Mater. Sci. Forum, 2002, 386–388, 299 CrossRef Search PubMed
- T. Murakami, C. N. Xu, A. Kitahara, M. Kawahara, Y. T. Akahashi, H. Inui and M. Yamaguchi, Intermetallics, 1999, 7, 1043 CrossRef CAS Search PubMed
- M. Senna, Solid State Ionics, 1993, 63–65, 3 CrossRef CAS Search PubMed
- J. Temuujin, Chem. Sustainable Dev., 2001, 9, 589 CAS Search PubMed
- E. Grigorova, T. S. Mandzhukova, M. Khristov, M. Yoncheva, R. Stoyanova and E. Zhecheva, J. Mater. Sci., 2011, 46, 7106 CrossRef CAS Search PubMed
- E. G. Avvakumov and L. G. Karakchiev, Chem. Sustainable Dev., 2004, 12, 287 CAS Search PubMed
- M. Senna, Int. J. Miner. Process., 1996, 44–45, 187 CrossRef CAS Search PubMed
- M. Senna, Y. Fujiwara, T. Isobe and J. Tanaka, Solid State Ionics, 2001, 141–142, 31 CrossRef CAS Search PubMed
- M. Senna, J. Eur. Ceram. Soc., 2005, 25, 1977 CrossRef CAS Search PubMed
- M. Senna, Cryst. Res. Technol., 1985, 20, 209 CrossRef CAS Search PubMed
- J. F. Liao and M. Senna, Mater. Res. Bull., 1995, 30, 385 CrossRef CAS Search PubMed
- T. Watanabe, T. Isobe and M. Senna, J. Solid State Chem., 1996, 122, 74 CrossRef CAS Search PubMed
- T. Watanabe, T. Isobe and M. Senna, J. Solid State Chem., 1997, 130, 284 CrossRef CAS Search PubMed
- T. Watanabe, T. Isobe and M. Senna, J. Solid State Chem., 1997, 122, 291 CrossRef Search PubMed
- T. Isobe, S. Komatsubara and M. Senna, J. Non-Cryst. Solids, 1992, 150, 144 CrossRef CAS Search PubMed
- K. Takeuchi, T. Isobe and M. Senna, J. Solid State Chem., 1994, 109, 401 CrossRef CAS Search PubMed
- J. G. Baek, T. Isobe and M. Senna, Solid State Ionics, 1996, 90, 269 CrossRef CAS Search PubMed
- J. G. Baek, T. Isobe and M. Senna, J. Am. Ceram. Soc., 1997, 80, 973 CrossRef CAS Search PubMed
- S. Shinohara, J. G. Baek, T. Isobe and M. Senna, J. Am. Ceram. Soc., 2000, 83, 3208 CrossRef CAS Search PubMed
- J. Ding, T. Tsuzuki and P. G. McCormick, J. Am. Ceram. Soc., 1996, 79, 2956 CrossRef CAS Search PubMed
- A. C. Dodd, Powder Technol., 2009, 196, 30 CrossRef CAS Search PubMed
- P. G. McCormick, T. Tsuzuki, J. S. Robinson and J. Ding, Adv. Mater., 2001, 13, 1008 CrossRef CAS Search PubMed
- F. Li, X. Yu, H. Pan, M. Wang and X. Xin, Solid State Sci., 2000, 2, 767 CrossRef CAS Search PubMed
- Y. X. Li, W. F. Chen, Z. Zhou, Z. Y. Gu and C. M. Chen, Mater. Lett., 2005, 59, 48 CrossRef CAS Search PubMed
- T. Tsuzuki and P. G. McCormick, Acta Mater., 2000, 48, 2795 CrossRef CAS Search PubMed
- P. Billik and G. Plesch, Scr. Mater., 2007, 56, 979 CrossRef CAS Search PubMed
- A. C. Dodd and P. G. McCormick, J. Eur. Ceram. Soc., 2002, 22, 1823 CrossRef CAS Search PubMed
- P. F. Xiao, M. O. Lai and L. Lu, Electrochim. Acta, 2012, 76, 185 CrossRef CAS Search PubMed
- Ü. Kersen and L. Holappa, Anal. Chim. Acta, 2006, 562, 110 CrossRef Search PubMed
- P. Billik and G. Plesch, Mater. Lett., 2007, 61, 1183 CrossRef CAS Search PubMed
- V. Kusigerski, M. Tadić, V. Spasojević, B. Antić, D. Marković, S. Boković and B. Matović, Scr. Mater., 2007, 56, 883 CrossRef CAS Search PubMed
- X. L. Cui and L. S. Cui, Key Eng. Mater., 2005, 280–283, 581 CrossRef CAS Search PubMed
- R. Yang, T. Xing, R. Xu and M. Li, Int. J. Refract. Met. Hard Mater., 2011, 29, 138 CrossRef CAS Search PubMed
- R. Yang, L. Cui, Y. Zheng and X. Cai, Mater. Lett., 2007, 61, 4815 CrossRef CAS Search PubMed
- P. Billik, M. Čaplovičová and L. Čaplovič, Mater. Res. Bull., 2010, 45, 621 CrossRef CAS Search PubMed
- T. Tsuzuki, F. Schäffel, M. Muroi and P. G. McCormick, Powder Technol., 2011, 210, 198 CrossRef CAS Search PubMed
- T. Kimura, in Advances in Ceramics – Synthesis and Characterization, Processing and Specific Applications, ed. C. Sikalidis, InTech, Rijeka, 2011, pp. 75–97 Search PubMed
-
(a) R. E. Treece, E. G. Gillan and R. B. Kanner, Comments Inorg. Chem., 1995, 16, 313 CrossRef CAS Search PubMed
- P. R. Bonneau, R. F. Jarvis and R. B. Kanner, Nature, 1991, 349, 510 CrossRef CAS Search PubMed
- G. B. Schaffer and P. G. McCormick, Scr. Metall., 1989, 23, 835 CrossRef CAS Search PubMed
- V. Hlavacek, Ceram. Bull., 1991, 70, 240 CAS Search PubMed
- P. Baláž, E. Godočíková, L. Kril'ová, P. Lobotka and E. Gock, Mater. Sci. Eng., A, 2004, 386, 442 Search PubMed
- P. Baláž, E. Godočíková, L. Takacs and E. Gock, Int. J. Mater. Prod. Technol., 2005, 23, 26 Search PubMed
- P. Baláž, A. Aláčová, E. Godočíková, J. Kováč, I. Škorvánek and J. Z. Jiang, Czech. J. Phys., 2004, 54, D197–D200 CrossRef Search PubMed
- P. Baláž, L. Takacs, J. Z. Jiang, V. Soika and M. Luxová, Mater. Sci. Forum, 2002, 386–388, 257 CrossRef Search PubMed
- P. Baláž, L. Takacs, M. Luxová, E. Godočíková and J. Ficeriová, Int. J. Miner. Process., 2004, 74, S365 CrossRef Search PubMed
- E. Gaffet, E. Bernard, J. C. Niepce, F. Charlot, C. Gras, G. Le Caër, J. L. Guichard, P. Delcroix, A. Mocellin and O. Tillement, J. Mater. Chem., 1999, 9, 305 RSC
- C. C. Koch, Mater. Sci. Technol., 1991, 15, 93 Search PubMed
- J. Ding, W. F. Miao, E. Pirault, R. Street and P. G. McCormick, J. Alloys Compd., 1998, 267, 199 CrossRef CAS Search PubMed
- T. Tsuzuki, J. Ding and P. G. McCormick, Phys. B, 1997, 239, 378 CrossRef CAS Search PubMed
- P. Matteazzi and G. Le Caër, Mater. Sci. Eng., A, 1992, 156, 229 CrossRef Search PubMed
- W. Zhuangzhi, W. Dezhi and S. Aokui, J. Cryst. Growth, 2010, 312, 340 CrossRef Search PubMed
- L.-D. Zhao, B.-P. Zhang, W.-S. Liu, H.-L. Zhang and J.-F. Li, J. Solid State Chem., 2008, 181, 3278 CrossRef CAS Search PubMed
- Z.-H. Ge, B.-P. Zhang, Y.-Q. Yu and P.-P. Shang, J. Alloys Compd., 2012, 514, 205 CrossRef CAS Search PubMed
- Y. Hwa, C.-M. Park and H.-J. Sohn, J. Electroanal. Chem., 2012, 667, 24 CrossRef CAS Search PubMed
- C. G. Tschakarov, G. G. Gospodinov and Z. Bontschev, J. Solid State Chem., 1982, 41, 244 CrossRef Search PubMed
- M. Atzmon, Phys. Rev. Lett., 1990, 64, 487 CrossRef CAS Search PubMed
-
(a) M. Zeghmati, E. Duverger and E. Gaffet, in Proceedings Cong. Canad. Mécan. Appl. CANCAM 95, ed. B. Tabarrock and S. Dost, 1995, vol. 2, p. 952 Search PubMed
- R. Ricceri and P. Matteazzi, J. Alloys Compd., 2003, 358, 71 CrossRef CAS Search PubMed
- Z. A. Munir, Ceram. Bull., 1988, 67, 342 CAS Search PubMed
- M. A. Bab and L. Mendoza-Zelis, Scr. Mater., 2004, 50, 99 CrossRef CAS Search PubMed
- G. B. Schaffer and P. G. McCormick, Metall. Trans. A, 1992, 23, 1285 CrossRef Search PubMed
- G. B. Schaffer and J. S. Forrester, J. Mater. Sci., 1997, 32, 3157 CrossRef CAS Search PubMed
- C. Deidda, F. Delogu, F. Maglia, U. Anselmi-Tamburini and G. Cocco, Mater. Sci. Eng., A, 2004, 375–377, 800 CrossRef Search PubMed
- K. Kudaka, K. Iizumi, T. Sasaki and H. Izumi, J. Am. Ceram. Soc., 2000, 83, 2887 CrossRef CAS Search PubMed
- A. Bakhshai, V. Soika, M. A. Susol and L. Takacs, J. Solid State Chem., 2000, 153, 371 CrossRef CAS Search PubMed
- L. Takacs and V. Šepelák, J. Mater. Sci., 2004, 39, 5487 CrossRef CAS Search PubMed
- F. Charlot, E. Gaffet, B. Zeghmati, F. Bernard and J.-C. Niepce, Mater. Sci. Eng., A, 1999, 262, 279 CrossRef Search PubMed
- F. Bernard, F. Charlot, E. Gaffet and J.-C. Niepce, Int. J. Self-Propag. High-Temp. Synth., 1998, 7, 233 CAS Search PubMed
- F. Charlot, E. Gaffet, F. Bernard, Ch. Gras and J.-C. Niepce, Mater. Sci. Forum, 1999, 312–314, 287 CrossRef CAS Search PubMed
- C. Suryanarayana, Mechanical Alloying and Milling, Marcel Dekker, New York, 2004 Search PubMed
- F. Charlot, C. Gras, M. Grammond, F. Bernard, E. Gaffet and J.-C. Niepce, J. Phys. Suppl. Coll., 1998, IV, 497 Search PubMed
- Ch. Gras, D. Vrel, E. Gaffet and F. Bernard, J. Alloys Compd., 2001, 314, 240 CrossRef CAS Search PubMed
- Ch. Gras, F. Charlot, F. Bernard, E. Gaffet and J.-C. Niepce, Acta Mater., 1999, 47, 2113 CrossRef CAS Search PubMed
- Ch. Gras, N. Bernsten, F. Bernard and E. Gaffet, Intermetallics, 2002, 10, 271 CrossRef CAS Search PubMed
- Ch. Gras, E. Gaffet, F. Bernard and J.-C. Niepce, Mater. Sci. Eng., A, 1999, 264, 94 CrossRef Search PubMed
- H. Shouha, E. Gaffet, F. Bernard and J.-C. Niepce, J. Mater. Sci. Eng. A, 2000, 35, 3221 Search PubMed
- F. Bernard, H. Souha and E. Gaffet, Mater. Sci. Eng., A, 2000, 284, 301 CrossRef Search PubMed
- H. Shouha, F. Bernard, E. Gaffet and B. Gillot, Thermochim. Acta, 2000, 351, 71 CrossRef Search PubMed
- V. Gauthier, F. Bernard, E. Gaffet, D. Vrel and J.-P. Larpin, Intermetallics, 2002, 10, 377 CrossRef CAS Search PubMed
- V. Gauthier, F. Bernard, E. Gaffet, C. Josse and J.-P. Larpin, Mater. Sci. Eng., A, 1999, 272, 334 CrossRef Search PubMed
- V. Gauthier, C. Josse, F. Bernard, E. Gaffet and J.-P. Larpin, Mater. Sci. Eng., A, 1999, 265, 117 CrossRef Search PubMed
-
(a) V. Gauthier, J.-P. Larpin, M. Vilasi, F. Bernard and E. Gaffet, Mater. Sci. Forum, 2001, 369–372, 793 CrossRef CAS Search PubMed
- R. M. L. Neto and C. J. Da Rocha, Key Eng. Mater., 2001, 189–191, 567 CrossRef Search PubMed
- K. Uenishi, T. Matsubara, M. Kambara and K. F. Kobayashi, Scr. Mater., 2001, 44, 2093 CrossRef CAS Search PubMed
- T. Matsubara, K. Uenishi and K. F. Kobayashi, Mater. Trans., JIM, 2000, 41, 631 CAS Search PubMed
- J. Lagerbom, T. Tiainen, M. Lehtonen and P. Lintula, J. Mater. Sci., 1999, 34, 1477 CrossRef CAS Search PubMed
- S. Murali, T. Sritharan and P. Hing, Int. J. Powder Metall., 2001, 37, 67 CAS Search PubMed
- J.-C. Niepce, F. Baras, F. Bernard, J.-P. Bonnet, S. Dubois, J.-C. Gachon, E. Gaffet, V. Gauthier, A. Lemarchand, R.-M. Marin-Ayral, T. Montesin, F. Nardou, M.-C. Record and D. Vrel, Int. J. Self-Propag. High-Temp. Synth., 2007, 16, 235 CrossRef CAS Search PubMed
- F. Bernard, S. Paris, D. Vrel, M. Gailhanou, J.-C. Gachon and E. Gaffet, Int. J. Self-Propag. High-Temp. Synth., 2002, 11, 181 CAS Search PubMed
- E. Gaffet, F. Charlot, D. Klein, F. Bernard and J. C. Niepce, Mater. Sci. Forum, 1998, 269–272, 379 CrossRef CAS Search PubMed
- F. Bernard, E. Gaffet, M. Gramond, M. Gailhanou and J. C. Gachon, J. Synchrotron Radiat., 2000, 7, 27 CrossRef CAS Search PubMed
- V. Gauthier, F. Bernard, E. Gaffet, C. Josse and J.-P. Larpin, Mater. Sci. Eng., A, 1999, 272, 334 CrossRef Search PubMed
- Ch. Gras, E. Gaffet, F. Bernard, F. Charlot and J.-C. Niepce, Mater. Sci. Forum, 1999, 312–314, 281 CrossRef CAS Search PubMed
- G. Cabouro, S. Chevalier, E. Gaffet, D. Vrel, N. Boudet and F. Bernard, Acta Mater., 2007, 55, 6051 CrossRef CAS Search PubMed
- G. Cabouro, S. Le Gallet, S. Chevalier, E. Gaffet, Y. Grin and F. Bernard, Powder Technol., 2011, 208, 526 CrossRef CAS Search PubMed
- F. Charlot, F. Bernard, D. Klein, E. Gaffet and J.-C. Niepce, Acta Mater., 1999, 47, 619 CrossRef CAS Search PubMed
- The Chemistry of Water, National Science Foundation (http://www.nsf.gov/news/special_reports/water/index_low.jsp).
- L. T. Alexander and M. G. Byers, J. Chem. Educ., 1932, 9, 916 CrossRef CAS Search PubMed
-
(a) G. A. Domrachev and D. A. Selivanovskii, Gorky: IMKh AN SSSR, 1990, 1, 20 Search PubMed
- A. D. Styrkas and N. G. Nikishina, High Energy Chem., 2007, 41, 396 CrossRef CAS Search PubMed
- W. Stumm, Processes at the Mineral-water and Particle-water Interfaces in Natural Systems, John Wiley & Sons Inc., New York, 1992 Search PubMed
- A. Z. Juhász, Part. Sci. Technol., 1998, 16, 145 CrossRef Search PubMed
- A. Z. Juhász and L. Opoczky, Mechanical Activation of Minerals by Grinding : Pulverizing and Morphology of Particles, Ellis Horwood Limited Chichester, Akademiai Kiado, Budapest, 1990 Search PubMed
- Y. X. Li, X. Z. Zhou, Y. Wang and X. Z. You, Mater. Lett., 2003, 58, 245 CrossRef Search PubMed
- P. Billik, M. Čaplovičová, T. Turányi, L. Čaplovič and B. Horváth, Mater. Res. Bull., 2011, 46, 2135 CrossRef CAS Search PubMed
- V. V. Boldyrev, Powder Technol., 2002, 122, 247 CrossRef CAS Search PubMed
- M. Fabián, M. Shopska, D. Paneva, G. Kadinov, N. Kostova, E. Turianicová, J. Briančin, I. Mitov, R. A. Kleiv and P. Baláž, Miner. Eng., 2010, 23, 616 CrossRef Search PubMed
- G. Baudet, V. Perrotel, A. Seron and M. Stellatelli, Powder Technol., 1999, 105, 125 CrossRef CAS Search PubMed
- P. Baláž, E. Turianicová, M. Fabián, R. A. Kleiv, J. Briančin and A. Obut, Int. J. Miner. Process., 2008, 88, 1 CrossRef Search PubMed
- V. Sydorchuk, S. Khalameida, V. Zazhigalov, J. Skubiszewska-Zieba, R. Leboda and K. Wieczorek-Ciurowa, Appl. Surf. Sci., 2010, 257, 446 CrossRef CAS Search PubMed
- F. Stenger, S. Mende, J. Schwedes and W. Peukert, Powder Technol., 2005, 156, 103 CrossRef CAS Search PubMed
- P. A. Rebinder, Z. Phys., 1931, 72, 191 CrossRef Search PubMed
- A. R. C. Westwod, J. S. Ahearn and J. J. Mills, Colloids Surf., 1981, 2, 1 CrossRef Search PubMed
- H. El-Shall and P. Somasundran, Powder Technol., 1984, 38, 274 Search PubMed
- P. Somasundaran and S. Shrotri, in Selected Topics in Mineral Processing, ed. R. Kumar and M. Pradip, Wiley Eastern Ltd, New Delhi, 1995, pp. 47–70 Search PubMed
- B. Kolláth and A. Z. Juhász, Cem. Concr. Res., 1996, 26, 1843 CrossRef Search PubMed
- A. I. Rusanov, N. E. Esipova and A. I. Emelina, Russ. J. Gen. Chem., 2007, 77, 2108 CrossRef CAS Search PubMed
- H. Ding, S. Lu and G. Du, Int. J. Miner., Metall. Mater., 2011, 18, 83 CrossRef CAS Search PubMed
- H. Ding, S. Lu, Y. Deg and G. Du, Trans. Nonferrous Met. Soc. China, 2007, 17, 1100 CrossRef CAS Search PubMed
- R. Kumar, S. Kumar, S. K. Badjena and S. P. Mehrotra, Metall. Mater. Trans. B, 2005, 36B, 473 Search PubMed
- T. C. Alex, R. Kumar, S. K. Roy and S. P. Mehrotra, Adv. Powder Technol., 2008, 19, 483 CAS Search PubMed
- V. M. Gun’ko, E. F. Voronin, L. V. Nosach, V. V. Turov, Z. Wang, A. P. Vasilenko, R. Leboda, J. Skubiszewska-Zieba, W. Janusz and S. V. Mikhalovsky, J. Colloid Interface Sci., 2011, 355, 300 CrossRef Search PubMed
- Y. Wang and E. Forssberg, China Particuol., 2007, 5, 193 CrossRef Search PubMed
- F. Stenger and W. Peukert, Chem. Eng. Technol., 2003, 26, 177 CrossRef CAS Search PubMed
- J. Liao and M. Senna, Solid State Ionics, 1993, 66, 313 CrossRef CAS Search PubMed
- M. Kitamura and M. Senna, Adv. Powder Technol., 2001, 12, 215 CrossRef CAS Search PubMed
- J. Kano, S. Saeki, F. Saito, M. Tanjo and S. Yamazaki, Int. J. Miner. Process., 2000, 60, 91 CrossRef CAS Search PubMed
- K. J. D. MacKenzie, J. Temuujin, M. E. Smith, P. Angerer and Y. Kameshima, Thermochim. Acta, 2000, 359, 87 CrossRef CAS Search PubMed
- A. Tonejc, M. Stubicar, A. M. Tonejc, K. Kosanovic, B. Subotic and I. Smit, J. Mater. Sci. Lett., 1994, 13, 519 CrossRef CAS Search PubMed
- T. Tsuchida and K. Horigome, Thermochim. Acta, 1995, 254, 359 CrossRef CAS Search PubMed
- T. C. Alex, R. Kumar, S. K. Roy and S. P. Mehrotra, Powder Technol., 2011, 208, 128 CrossRef CAS Search PubMed
- A. Drief and F. Nieto, Clays Clay Miner., 1999, 47, 417 CAS Search PubMed
- R. L. Frost, E. Makó, J. Kristóf, E. Horvath and J. T. Kloprogge, J. Colloid Interface Sci., 2001, 239, 458 CrossRef CAS Search PubMed
- R. L. Frost, E. Makó, J. Kristóf, E. Horvath and J. T. Kloprogge, Langmuir, 2001, 17, 4731 CrossRef CAS Search PubMed
- E. Mendelovici, J. Mater. Sci. Lett., 2001, 20, 81 CrossRef CAS Search PubMed
- J. Kameda, K. Saruwatari and H. J. Tanaka, J. Colloid Interface Sci., 2004, 275, 225 CrossRef CAS Search PubMed
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2928 CrossRef Search PubMed
- F. Dellisanti, V. Minguzzi and G. Valdrè, Appl. Clay Sci., 2006, 31, 282 CrossRef CAS Search PubMed
- F. Dellisantia and G. Valdrèb, Int. J. Miner. Process., 2012, 102–103, 69 CrossRef Search PubMed
- A. Tang, L. Su and C. Li, Powder Technol., 2012, 218, 86 CrossRef CAS Search PubMed
- G. Greifzu, R. Kumar and T. C. Alex, Mechanically induced reactivity of hydrothermally prepared boehmite (γ-AlOOH), Report of Investigation OLP-0177, CSIR-National Metallurgical Laboratory, Jamshedpur, 2012, pp. 1–29 Search PubMed
- R. Kiriyama, Y. Tamai and F. Kanamura, Nippon Kagaku Zassi, 1967, 88, 8 CrossRef Search PubMed
- V. V. Boldyrev, Kinet. Catal., 1972, 13, 1411 CAS Search PubMed
- N. V. Kosova, A. Kh. Khabibullin and V. V. Boldyrev, Solid State Ionics, 1997, 101–103, 53 CAS Search PubMed
- K. Hamada and M. Senna, J. Mater. Sci., 1996, 31, 1725 CrossRef CAS Search PubMed
- M. Yoshimura, W. L. Suchanek and K. Byrappa, MRS Bull., 2000, 25, 17 CrossRef CAS Search PubMed
- J. Temuujin, Chem. Sustainable Dev., 2001, 9, 589 CAS Search PubMed
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117 CrossRef CAS Search PubMed
- F. Saito, G. Mi and M. Hanada, Solid State Ionics, 1997, 101–103, 37 CAS Search PubMed
- K. Wieczorek-Ciurowa and K. Gamrat, Mater. Sci.-Pol., 2007, 25, 219 CAS Search PubMed
-
(a) J. Temuujin, K. Okada and K. Mackenzie, J. Am. Ceram. Soc., 1998, 81, 754 CrossRef CAS Search PubMed
- J. Liao, K. Hamada and M. Senna, J. Mater. Synth. Process., 2000, 8, 305 CrossRef CAS Search PubMed
-
(a) R. E. Riman, W. L. Suchanek and M. M. Lencka, Ann. Chim. Sci. Mater., 2002, 27, 15 CrossRef CAS Search PubMed
-
(a) W. L. Suchanek, P. Shuk, K. Byrappa, R. E. Riman, K. S. TenHuisen and V. F. Janas, Biomaterials, 2002, 23, 699 CrossRef CAS Search PubMed
- A. Fahami, B. Nasiri-Tabrizi and R. Ebrahimi-Kahrizsangi, Ceram. Int., 2012, 38, 6729 CrossRef CAS Search PubMed
- C. R. Nelson, S. Olson, S. A. Poling and S. W. Martin, Chem. Mater., 2006, 18, 6436 CrossRef CAS Search PubMed
- H. El Briak-BenAbdeslam, M. P. Ginebra, M. Vert and P. Boudeville, Acta Biomater., 2008, 4, 378 CrossRef CAS Search PubMed
- C. W. Chen, R. E. Riman, K. S. TenHuisen and K. Brown, J. Cryst. Growth, 2004, 270, 615 CrossRef CAS Search PubMed
- E. Avvakumov, E. Devyatkina and N. Kosova, J. Solid State Chem., 1994, 113, 379 CrossRef CAS Search PubMed
- T. Friščić and W. Jones, in Frontiers in Mechanochemistry and Mechanical Alloying, ed. R. Kumar, S. Srikanth and S. P. Mehrotra, CSIR-National Metallurgical Laboratory, Jamshedpur, 2011, pp. 31–40 Search PubMed
-
(a) T. Iwasaki, K. Kosaka, T. Yabuuchi, S. Watano, T. Yanagida and T. Kawai, Adv. Powder Technol., 2009, 20, 521 CrossRef CAS Search PubMed
- T. Iwasaki, N. Sato, K. Kosaka, S. Watano, T. Yanagida and T. Kawai, J. Alloys Compd., 2011, 509, L34 CrossRef CAS Search PubMed
- R. Kumar, S. Kumar, T. C. Alex, S. Srikanth and S. P. Mehrotra, in Experimental and Theoretical Approaches to Modern Mechanochemistry, ed. G. Mulas and F. Delogu, Transworld Research Network, Chennai, 2010, pp. 255–272 Search PubMed
- S. P. Mehrotra, Trans. Indian Inst. Met., 2007, 60, 1 CAS Search PubMed
- S. Mende, G. Kolb and U. Enderle, Nanoparticle: Grinding and Dispersing, www.ceramicindustry.com, 2006, p. 18 Search PubMed
- A. Kwade and J. Schwedes, Powder Technol., 2002, 122, 109 CrossRef CAS Search PubMed
- C. M. Palmer and G. D. Johnson, JOM, 2005, 57, 40 CrossRef CAS Search PubMed
-
(a) S. Kumar, R. Kumar and A. Bandopadhyay, Resour., Conserv. Recycl., 2006, 48, 301 CrossRef Search PubMed
- http://www.isamill.com/EN/Downloads/Latest%20News/NEWS%20RELEASE%20-%20100th%20IsaMill%2016-12-11.pdf.
- K. Raj and L. M. Aziz, US Patent, 5958282, 1999 Search PubMed
- W. Peukert, H. C. Schwarzer and F. Stenger, Chem. Eng. Process., 2005, 44, 245 CrossRef CAS Search PubMed
- F. Stenger, M. Götzinger, P. Jakob and W. Peukert, Part. Part. Syst. Charact., 2004, 21, 31 CrossRef CAS Search PubMed
- E. Bilgilia, R. Hamey and B. Scarlett, Chem. Eng. Sci., 2006, 61, 149 CrossRef Search PubMed
- C. Frances, N. Le Bolay, K. Belaroui and M. N. Pons, Int. J. Miner. Process., 2001, 61, 41 CrossRef CAS Search PubMed
- R. Kumar, T. C. Alex, J. P. Srivastava, B. R. Kumar, Z. H. Khan, S. P. Mahapatra and C. R. Mishra, Met., Mater. Processes, 2004, 16, 171 CAS Search PubMed
- T. C. Alex, Mechanical activation and reactivity of aluminium oxyhydroxide minerals, PhD Thesis, Indian Institute of Technology, Kanpur, 2012 Search PubMed
- B. Fritsch, Ber. Dtsch. Keram. Ges., 1963, 42, 149 Search PubMed
- K. Okada, A. Kuriki, S. Hayashi, T. Yano and N. Otsuka, J. Mater. Sci. Lett., 1993, 12, 862 CrossRef CAS Search PubMed
- S. Kikuchi, T. Ban, K. Okada and N. Otsuka, J. Mater. Sci. Lett., 1992, 11, 471 CrossRef CAS Search PubMed
- R. Oberacker, S. Poehnitzsch and H. Hofius, Ceram. Forum Int. Yearb., 2001, 78, 45 Search PubMed
- H. Ding, S. Lu, Y. Deg and G. Du, Trans. Nonferrous Met. Soc. China, 2007, 17, 1100 CrossRef CAS Search PubMed
- L. Turčániová and P. Baláž, J. Mater. Synth. Process., 2000, 8, 1064 Search PubMed
- F. Pawlek, M. J. Kheiri and R. Kammel, in Light Metals, ed. E. R. Cutshall, The Minerals, Metals & Materials Society, Warrendale, 1992, pp. 1–95 Search PubMed
- P. G. Mc Cormick, T. Picaro and P. A. I. Smith, Miner. Eng., 2002, 15, 211 CrossRef CAS Search PubMed
-
(a) L. G. Shumskaya, J. Min. Sci., 2002, 38, 299 CrossRef Search PubMed
- R. Kumar, T. C. Alex, M. K. Jha, Z. H. Khan, S. P. Mahapatra and C. R. Mishra, in Light Metals, ed. P. Crepeau, The Minerals, Metals & Materials Society, Warrendale, 2004, pp. 31–34 Search PubMed
- R. Kumar, T. C. Alex, Z. H. Khan, S. P. Mahapatra and S. P. Mehrotra, in Light Metals, ed. H. Kvande, The Minerals, Metals & Materials Society, Warrendale, 2005, pp. 77–79 Search PubMed
-
(a) S. Fortin and G. Forté, in Light Metals, ed. M. Sorlie, The Minerals, Metals & Materials Society, Warrendale, 2007, pp. 87–92 Search PubMed
- T. Picaro, Red mud processing, Australian Patent 719126, 1997 Search PubMed
- S. Song and H. M. Jennings, Cem. Concr. Res., 1999, 29, 159 CrossRef CAS Search PubMed
- C. Bernhardt, E. Reinsch and K. Husemann, Powder Technol., 1999, 105, 357 CrossRef CAS Search PubMed
- F. Stenger, S. Mende, J. Schwedes and W. Peukert, Powder Technol., 2005, 156, 103 CrossRef CAS Search PubMed
- J. J. Gilman, Science, 1996, 274, 65 CrossRef CAS Search PubMed
- T. Luty, P. Orden and C. J. Eckhardt, J. Chem. Phys., 2002, 117, 1775 CrossRef CAS Search PubMed
- K. Tkáčová, Mechanical Activation of Minerals, Elsevier, Amsterdam, 1989 Search PubMed
- B. M. Rosen and V. Percec, Nature, 2007, 446, 381 CrossRef CAS Search PubMed
- E. V. Boldyreva, in Frontiers in Mechanochemistry and Mechanical Alloying, ed. R. Kumar, S. Srikanth and S. P. Mehrotra, Jamshedpur, 2011, pp. 17–30 Search PubMed
-
(a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC
- H. Watanabe and M. Senna, Tetrahedron Lett., 2005, 46, 6815 CrossRef CAS Search PubMed
- H. Watanabe and M. Senna, Tetrahedron Lett., 2006, 47, 4481 CrossRef CAS Search PubMed
- M. Senna, Process. Appl. Ceram., 2010, 4, 183 CAS Search PubMed
- T. Watanabe, N. Wakiyama, A. Kasai and M. Senna, Ann. Chem. Sci. Mater., 2004, 29, 53 CrossRef CAS Search PubMed
- M. Senna, V. Šepelák, J. Shi, B. Bauer, A. Feldhoff, V. Laporte and K.-D. Becker, J. Solid State Chem., 2012, 187, 51 CrossRef CAS Search PubMed
- R. Yanagawa, C. Ando, H. Chazono, H. Kishi and M. Senna, J. Am. Ceram. Soc., 2007, 90, 809 CrossRef CAS Search PubMed
- K. Oguchi, C. Ando, H. Chazono, H. Kish and M. Senna, J. Phys. IV, 2005, 128, 33 Search PubMed
- T. Yamamoto, H. Hiori and H. Moriwake, Jpn. J. Appl. Phys., 2000, 39, 5683 CrossRef CAS Search PubMed
- C. Ando, K. Tsuzuku, T. Kobayashi, H. Kishi, S. Kuroda and M. Senna, J. Mater. Sci.: Mater. Electron., 2009, 20, 844 CrossRef CAS Search PubMed
- E. Matsui, Y. Abe, M. Senna, A. Guerfi and K. Zaghib, J. Am. Ceram. Soc., 2008, 91, 1522 CrossRef CAS Search PubMed
- X. Michelet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef Search PubMed
- T. Friščić, J. Mater. Chem., 2010, 20, 7599 RSC
- V. M. Kanevskii, Crystallogr. Rep., 2011, 56, 662 CrossRef CAS Search PubMed
- V. Šepelák, M. Myndyk, M. Fabián, K. L. DaSilva, A. Feldhoff, D. Menzel, M. Ghafari, H. Hahn, P. Heitjans and K. D. Becker, Chem. Commun., 2012, 48, 11121 RSC
- A. V. Firth, S. W. Haggata, P. K. Khanna, S. J. Williams, J. W. Allen, S. W. Magennis, I. D. W. Samuel and D. J. Cole-Hamilton, J. Lumin., 2004, 109, 163 CAS Search PubMed
- L. Wang, D. K. Nagesha, S. Selvarasah, M. R. Dokmeci and R. L. Carrier, J. Nanobiotechnol., 2008, 6, 11 CrossRef Search PubMed
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468 CrossRef CAS Search PubMed
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11 CrossRef CAS Search PubMed
- I. L. Medinitz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef Search PubMed
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 49, 122 Search PubMed
- A. R. Clapp, E. R. Goldman and H. Mattoussi, Nat. Protoc., 2006, 1, 1258 CrossRef CAS Search PubMed
- H.-Y. Xie, J.-G. Liang, Y. Liu, Z.-L. Zhang, D.-W. Peng, Z.-K. He, Z.-X. Lu and W.-H. Huang, J. Nanosci. Nanotechnol., 2005, 5, 880 CrossRef CAS Search PubMed
- G. L. Tan, J. H. Du and Q. J. Zhang, J. Alloys Compd., 2009, 468, 421 CrossRef CAS Search PubMed
-
(a) P. Baláž, R. Jardin, E. Dutková, M. J. Sayagués, M. Baláž, G. Mojžišová, J. Mojžiš, E. Turianicová and M. Fabián, Acta Phys. Pol., A, 2012, 122, 224 Search PubMed
- O. I. Lomovskij, Chem. Sustainable Dev., 1994, 2, 473 Search PubMed
- O. I. Lomovskij and V. V. Boldyrev, Mechanochemistry for Solving Environmental Problems, GPNTB SO RAN, Novosibirsk, 2006 (in Russian) Search PubMed
- V. V. Boldyrev and K. Tkáčová, J. Mater. Synth. Process., 2000, 8, 121 CrossRef CAS Search PubMed
- V. V. Boldyrev, Russ. Chem. Rev., 2006, 75, 177–189 CrossRef CAS Search PubMed
- C. Suryanarayana, E. Ivanov and V. V. Boldyrev, Mater. Sci. Eng., A, 2001, 304–306, 151 CrossRef Search PubMed
- C. Suryanarayana, Non-equilibrium Processing of Materials, Pergamon Press, Oxford, 1999 Search PubMed
- Proceedings of the 8th European Symposium on Martensitic Transformation ESOMAT, ed. F. Neves, F. M. Braz Fernandes, I. Martins, J. B. Correia, M. Oliveira, E. Gaffet, T.-Y. Wang, M. Lattemann, J. Suffner and H. Hahn, Prague, 2009 Search PubMed
- F. Neves, I. Martins, J. B. Correia, M. Oliveira and E. Gaffet, Mater. Sci. Eng., A, 2008, 473, 336 CrossRef Search PubMed
- F. Neves, I. Martins, J. B. Correia, M. Oliveira and E. Gaffet, Intermetallics, 2007, 15, 1623 CrossRef CAS Search PubMed
- F. Neves, F. M. Braz Fernandes, I. Martins, J. B. Correia, M. Oliveira, E. Gaffet, T.-Y. Wang, M. Lattemann, J. Suffner and H. Hahn, Smart Mater. Struct., 2009, 18, 115003 CrossRef Search PubMed
- F. Neves, I. Martins, J. B. Correia, M. Oliveira and E. Gaffet, Intermetallics, 2008, 16, 889 CrossRef CAS Search PubMed
- F. Bernard, S. Le Gallet, N. Spinassou, S. Paris, E. Gaffet, J. N. Woolman and Z. A. Munir, Sci. Sintering, 2004, 36, 155 CrossRef CAS Search PubMed
- Z. A. Munir, F. Charlot, E. Gaffet and F. Bernard, US Patent, Canadian/Japanese/European Patent, 6200515, WO0112366, 2001 Search PubMed
- G. Cabouro, S. Chevalier, E. Gaffet, Y. Grin and F. Bernard, J. Alloys Compd., 2008, 465, 344 CrossRef CAS Search PubMed
- Ch. Gras, F. Bernard, F. Charlot, E. Gaffet and Z. A. Munir, J. Mater. Res., 2002, 13, 542 CrossRef Search PubMed
- S. Paris, C. Pighini, E. Gaffet, Z. A. Munir and F. Bernard, Int. J. Self-Propag. High-Temp. Synth., 2008, 17, 183 CrossRef CAS Search PubMed
- G. Ji, Th. Grosdidier, F. Bernard, S. Paris, E. Gaffet and S. Launois, J. Alloys Compd., 2007, 434–435, 358 CrossRef CAS Search PubMed
- Th. Grosdidier, G. Ji, F. Bernard, E. Gaffet, Z. A. Munir and S. Launois, Intermetallics, 2006, 14, 1208 CrossRef CAS Search PubMed
- G. Ji, D. Goran, F. Bernard, T. Grosdidier, E. Gaffet and Z. A. Munir, J. Alloys Compd., 2006, 420, 158 CrossRef CAS Search PubMed
- S. Paris, E. Gaffet, F. Bernard and Z. A. Munir, Scr. Mater., 2004, 50, 691 CrossRef CAS Search PubMed
- S. Paris, C. Pignini, S. Chevalier, O. El Kedim, E. Gaffet, Z. A. Munir and F. Bernard, Int. J. Self-Propag. High-Temp. Synth., 2003, 12, 137 CAS Search PubMed
- S. Paris, Ch. Valot, L. Gosmain, E. Gaffet, F. Bernard and Z. A. Munir, J. Mater. Res., 2003, 18, 2331 CrossRef CAS Search PubMed
- F. Charlot, E. Gaffet, F. Bernard and Z. A. Munir, J. Am. Ceram. Soc., 2001, 84, 910 CrossRef Search PubMed
- O. El Kedim, S. Paris, C. Phigini, F. Bernard, E. Gaffet and Z. A. Munir, Mater. Sci. Eng., A, 2004, 369, 49 CrossRef Search PubMed
- V. Gauthier, F. Bernard, E. Gaffet, Z. Munir and J.-P. Larpin, Intermetallics, 2001, 9, 571 CrossRef CAS Search PubMed
- T. E. Madey, J. Y. Yates, Jr., D. R. Sandstrom and R. J. H. Voorhoeve, in Treatise on Solid State Chemistry, Surfaces II, Plenum Press, New York, 1976, vol. 6B, pp. 1–124 Search PubMed
- Annual Report 2009–2010, Department of Science and Technology, Ministry of Science and Technology, New Delhi, India.
-
(a) Perspectives in Catalysis: A Chemistry for the 21st Century, ed. J. M. Thomas and K. O. Zamaraev, Oxford, Blackwell Science, 1992 Search PubMed
-
(a) F. Miani and F. Maurigh, in Encyclopaedia of Nanoscience and Nanotechnology, Marcel Dekker, New York, 2004, p. 1787–1795 Search PubMed
- R. A. Buyanov, V. V. Molchanov and V. V. Boldyrev, Catal. Today, 2009, 144, 212 CrossRef CAS Search PubMed
-
(a) R. A. Buyanov, V. V. Molchanov and V. V. Boldyrev, Hosokawa Powder Technology Foundation, KONA Powder Part. J., 2009, 27, 38 CAS Search PubMed
-
(a) V. V. Molchanov and R. A. Buyanov, Kinet. Catal., 2001, 42, 366 CrossRef CAS Search PubMed
-
(a) P. Jessop, Green Chem., 2011, 13, 1391 RSC
-
(a) I. Mitov, D. Dotcheva, Z. Cherkezova-Zheleva and V. Mitrov, in Metal-Ligand Interactions in Chemistry, Physics and Biology, ed. N. Russo and D. Salahub, NATO Sci. Series, Miesto, 2000, vol. 546, p. 383 Search PubMed
- H. Castricum, H. Bakker and E. Poels, Mater. Sci. Eng., A, 2001, 304–306, 418 CrossRef Search PubMed
- V. Zazhigalov, J. Haber, J. Stoch, L. Bogutskaya and I. Bacherikova, Stud. Surf. Sci. Catal., 1996, 101, 1039 CrossRef CAS Search PubMed
- V. Zazhigalov, J. Haber, J. Stoch, A. Kharlamov, I. Bacherikova and A. Kowal, Solid State Ionics, 1997, 101–103, 1257 CrossRef CAS Search PubMed
- V. A. Zazhigalov, S. V. Khalameida, N. S. Litvin, I. V. Bacherikova, J. Stoch and L. Depero, Kinet. Catal., 2008, 49, 692 CrossRef CAS Search PubMed
- V. P. Isupov, L. Chupakhina and R. Mitrofanova, J. Mater. Synth. Process., 2000, 8, 251 CrossRef CAS Search PubMed
- X. Wang, L. He, Y. He, J. Zhang and C.-Y. Su, Inorg. Chim. Acta, 2009, 362, 3513 CrossRef CAS Search PubMed
- U. Kamolphop, S. Taylor, J. Breen, R. Burch, J. Delgado, S. Chansai, C. Hardacre, S. Hengrasmee and S. James, ACS Catal., 2011, 1, 1257 CrossRef CAS Search PubMed
- J. Paulusse and R. Sijbesma, Chem. Commun., 2008, 4416 RSC
-
(a) R. Schrader, Freiberg. Forschungsh. A, 1966, 392, 81 CAS Search PubMed
- J. Eckell, Z. Elektrochem., 1933, 39, 433 CAS Search PubMed
- L. E. Craatty and A. V. Granato, J. Chem. Phys., 1957, 26, 96 CrossRef Search PubMed
- A. Krause and A. Herman, Rocnicki Chem., 1958, 32, 1025 CAS Search PubMed
- K. B. Keating, A. G. Rozner and J. L. Youngblood, J. Catal., 1965, 4, 608 CrossRef CAS Search PubMed
- I. Uhara, S. Yanagimoto, K. Tani and G. Adachi, Nature, 1961, 192, 867 CrossRef Search PubMed
- S. Kishimoto, J. Phys. Chem., 1962, 66, 2694 CrossRef CAS Search PubMed
- M. J. Diánez, J. Carrión and J. M. Criado, Proc. 1st International Conference on Mechanochemistry INCOME 93, Cambridge Interscience Publishing, Cambridge UK, 1993, p. 153 Search PubMed .
- J. M. Criado and M. J. Diánez, J. Mater. Sci., 1991, 26, 821 CrossRef CAS Search PubMed
- K. Wieczorek-Ciurowa, in High-energy ball milling, ed. M. Sopicka-Lizer, Woodhead Publishing Ltd., Cambridge UK, 2010, pp. 193–223 Search PubMed
- K. Wieczorek-Ciurowa, D. Oleszak and K. Gamrat, Chem. Sustainable Dev., 2007, 15, 255 CAS Search PubMed
- J. Rakoczy, J. Nizioł, K. Wieczorek-Ciurowa and P. Dulian, React. Kinet., Mech. Catal., 2013, 108, 81 CrossRef CAS Search PubMed
- R. Mamgbi, J. Nizioł, J. Rakoczy and K. Wieczorek-Ciurowa, Book of Abstracts 10th International Conference Solid State Chemistry, Pardubice, 2012, p. 86 Search PubMed
- J. Nizioł, J. Rakoczy and K. Wieczorek-Ciurowa, Proceedings of International Symposium on Alternative Clean Synthetic Fuels, Munich, 2012, 306 Search PubMed .
- K.-S. Lin, S. Chowdhury, H.-P. Yeh, W.-T. Hong and Ch.-T. Yeh, Catal. Today, 2011, 164, 251 CrossRef CAS Search PubMed
- Y. Taufiq-Yap, Y. Wong, Y. Kamiya and W. Tang, J. Nat. Gas Chem., 2008, 17, 232 CrossRef CAS Search PubMed
- A. Sokolov, D.-K. Bučar, J. Baltrusaitis, S. Gu and L. MacGillivray, Angew. Chem., Int. Ed., 2010, 49, 4273 CrossRef CAS Search PubMed
-
(a) T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66 CrossRef Search PubMed
-
(a) S. Hick, C. Griebel, D. Restrepo, J. Truitt, E. Buker, C. Bylda and R. Blair, Green Chem., 2010, 12, 468 RSC
-
(a) J. Gimzewski and C. Joachim, Science, 1999, 283, 1683 CrossRef CAS Search PubMed
- F. Habashi, A Textbook of Hydrometallurgy, Metallurgie Extractive Quebec, Sainte Foy, 1993 Search PubMed
-
(a) I. J. Corrans and J. E. Angove, Miner. Eng., 1991, 4, 763 CrossRef Search PubMed
- H. E. Evans and G. D. Johnson, in Copper Hydrometallurgy Roundtable ′99, Randol International, Phoenix, 1999 Search PubMed
- www.isamill.com.
- P. Baláž, F. Sekula, F. Jusko, M. Kočí, V. Dugas and L. Lauko, Slovak Patent, 081-94, 1994 Search PubMed
- P. Baláž and E. Dutková, in Frontiers in Mechanochemistry and Mechanical Alloying, ed. R. Kumar, S. Srikanth and S. P. Mehrotra, National Metallurgical Laboratory, Jamshedpur, 2011, pp. 229–239 Search PubMed
- P. Baláž and M. Achimovičová, Hydrometallurgy, 2006, 84, 60 CrossRef Search PubMed
- C. G. Anderson and S. M. Nordwick, in Proceedings of EPD Congress, ed. G. W. Warren, Anaheim, 1996, pp. 323–341 Search PubMed
- P. Baláž, F. Sekula, Š. Jakabský and R. Kammel, Miner. Eng., 1995, 8, 1299 CrossRef Search PubMed
- P. Baláž, R. Kammel, F. Sekula and Š. Jakabský, in Proceedings of 20th International Mineral Processing Congress, ed. H. Hoberg and H. von Blottnitz, Aachen, 1997, vol. 4, pp. 149–159 Search PubMed
- P. Baláž, R. Kammel, M. Kušnierová and M. Achimovičová, in Proceedings of International Conference Hydrometallurgy ’94, Chapman and Hall, London, 1994, pp. 209–218 Search PubMed
Footnote |
| † Dedicated to the memory of Prof. Pavel Yurievich Butyagin. |
| This journal is © The Royal Society of Chemistry 2013 |